 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Recombinant Expression and Characterization of N-terminal 43-amino Acid Deleted Human Apolipoprotein A-I (apoA-IΔ43) and Reconstitution of High-Density Lipoprotein Nanoparticle
Niphawan Panti, Sasithorn Sirilun, Jakkaphan Kumsab, Yodying Yingchutrakul, and Kasirawat Sawangrat*Published Date : September 19, 2025
DOI : https://doi.org/10.12982/NLSC.2025.074
Journal Issues : Number 4, October-December 2025
Abstract High-density lipoproteins (HDL) play crucial roles in cholesterol homeostasis and have emerged as promising therapeutic targets and drug delivery vehicles. This study developed an optimized protocol for recombinant expression of N-terminal 43-amino acid deleted human apolipoprotein A-I (apoA-I∆43) in Escherichia coli BL21(DE3) and subsequent reconstitution of HDL-mimetic nanoparticles. Expression optimization revealed that an induction temperature of 37°C, a 3-hour duration, and IPTG concentration of 0.1 mM yielded the highest apoA-I∆43 expression level (~20 mg/L culture). The purified apoA-I∆43 was characterized by LC-MS/MS, confirming a 185-amino acids of the expected 265-amino acid sequence. Reconstituted HDL-mimetic (rHDL) nanoparticles were successfully prepared using apoA-I∆43 and distearoyl-phosphatidylcholine (DSPC). These rHDL nanoparticles exhibited HDL-specific characteristics such as density (1.063-1.21 g/mL) and hydrodynamic diameter (11.7 ± 3.4 nm). This protocol provides a reliable platform for producing apoA-I∆43 for pharmaceutical applications and drug delivery system development.
Keywords: Apolipoprotein A-I, Recombinant expression, High-density lipoprotein, Nanoparticles, Drug delivery vehicles
Funding: This research was supported by Chiang Mai University Junior Research Fellowship Program (JRCMU2564_013) and Faculty of Pharmacy Research Grant for New Scholar 2564.
Citation: Panti, N., Sirilun, S., Kumsab, J., Yingchutrakul, Y., and Sawangrat, K. 2025. Recombinant expression and characterization of N-terminal 43-amino acid deleted human apolipoprotein A-I (apoA-IΔ43) and reconstitution of high-density lipoprotein nanoparticle. Natural and Life Sciences Communications. 24(4): e2025074.
INTRODUCTION
High-density lipoproteins (HDL) play a crucial role in cholesterol homeostasis and have emerged as promising therapeutic targets and drug delivery vehicles (Lerch et al., 1996; Ben-Aicha et al., 2020; van der Vorst, 2020; Fox et al., 2021). It represents a group of lipoproteins with an average diameter of 8-10 nm and density of 1.063-1.21 g/mL (Havel et al., 1955). In clinical studies, plasma HDL level demonstrates an inverse correlation with the risk of coronary artery disease (van der Vorst, 2020). This natural nanoparticle not only removes excess cholesterol from peripheral tissues but also exhibits promising attributes such as anti-inflammatory activities (Navab et al., 2004; Gordon et al., 2011; Cochran et al., 2021; AbdelHafez, 2022). Apolipoprotein A-I (apoA-I), the main protein component of HDL, is a 243-amino acid polypeptide with molecular mass of approximately 28,300 Da (van der Vorst, 2020). ApoA-I is synthesized by liver (70%) and small intestine (30%) in lipid-free form (van der Vorst, 2020). In plasma, apoA-I interacts with ATP-binding cassette transporter A1 (ABCA1) to retrieve phospholipids and cholesterol from peripheral cells, forming a small, discoidal-shaped HDL or nanodisc (van der Vorst, 2020).
Recently, functions of HDL other than anti-atherosclerosis activities have been explored. Reports have suggested that rHDL can be synthesized in vitro by the interaction of apoA-I and phospholipids (Jonas, 1986; Fukuda et al., 2020), can be used as a drug delivery vehicle (Ryan, 2008; Zhang et al., 2015; Suda et al., 2017; Fox et al., 2021; Deuringer et al., 2022). For example, an rHDL construct made from an apoA-I mutant which has C-terminal fusion with cell penetrating peptide (penetratin) showed higher cellular uptake compared to an rHDL construct without Penetratin (Suda et al., 2017). Moreover, rHDL bearing cell-penetrating peptide and neovascular targeting peptide showed more than 10-fold improvement in its anti-neovasculature activities (Fukuda et al., 2023). These findings highlight the versatility and importance of apoA-I in pharmaceutical applications.
Obtaining apoA-I represents a critical bottleneck in rHDL reconstitution which usually takes more time and resources than rHDL reconstitution itself (Fukuda et al., 2020). Recombinant protein technology offers a solution, enabling large-scale apoA-I production by competent cells such as Escherichia coli BL21(DE3) followed by histidine-tagged affinity chromatography. Previous studies have shown that N-terminal 43-amino acid deleted apoA-I (apoA-I∆43) maintains similar lipid-binding properties and exhibits only slightly reduced lecithin cholesterol acyltransferase (LCAT) activation compared to full-length native apoA-I. This truncated variant offers advantages in recombinant expression while preserving essential biological functions (Borhani et al., 1997; Rogers et al., 1997), making it a suitable candidate for our rHDL reconstitution.
This study developed a complete protocol for apoA-I∆43 synthesis. Since expression conditions such as temperature, culture time, and Isopropyl-ß-D-thiogalactopyranoside (IPTG) concentration significantly affect the amount and quality of target protein production, the optimal expression conditions need to be determined. The obtained apoA-I∆43 was purified and sequence-characterized by LC-MS/MS. In addition, pilot-scale reconstitution of rHDL was performed and characterized. The goal is to establish the protocol for apoA-I∆43 synthesis for further use in pharmaceutical applications.
MATERIALS AND METHODS
Materials
Bacterial expression of apoA-I∆43 mutant and optimization of the expression condition
The native human apoA-I amino acid sequence was obtained from GenPept (accession ID:3K2S_A). The cDNA encoding the apoA-IΔ43 mutant was synthesized and cloned into pET100/D-topo protein expression vector (Invitrogen®). The proposed amino acid sequence is shown in Figure 1. The obtained plasmid vector was transformed into One Shot™ BL21 Star™ (DE3) chemically competent E. coli (Invitrogen®) using heat shock protocol as described in our previous report with some modifications (Panti et al., 2021). The transformant was plated into Luria Bertani agar containing 50 µg/mL ampicillin as a selection agent. After a successful transformation, a single colony was transferred into Luria Bertani broth containing 50 µg/mL ampicillin without lactose to start preculture. This preculture was allowed to grow overnight in the incubator shaker at 37°C and a rotational speed of 140 rpm. Later, this preculture was expanded into mass culture vessel. This mass culture was allowed to grow in the incubator/shaker until the 600 nm optical density (OD600) reached 0.4-0.6. After the target OD600 was achieved, the mass culture was split into smaller culture tubes that were subjected to different induction conditions. Three variable factors of the induction condition (IPTG concentration, temperature during IPTG induction, and time after IPTG induction) were investigated. At a predetermined interval, cell pellets from 500 µL of crude culture media were collected. The samples were kept at -80°C until analyzed by SDS-PAGE.

Figure 1. Proposed amino acid sequence of apoA-IΔ43 mutant. Underlined characters represent terminal amino acids derived from pET100/D-topo vector. The highlighted represents the amino acids that are observed in LC-MS protein sequencing.
SDS-PAGE analysis
Polyacrylamide gel electrophoresis was used to determine the expression of the target protein. We manually cast polyacrylamide gel using Bio-Rad gel casting kit (1.0 mm thickness, 10-well comb). The samples were mixed with Laemmli 2X SDS-PAGE sample buffer (Bio-Rad, CA, USA), heated at 95°C for 5 min. and cooled to room temperature before being loaded into the gel. The electrophoresis was performed in a Mini-PROTEAN® system (Bio-Rad CA, USA). After that, the gel was fixed and stained with COOMASSIE nano® Protein Staining Solution (Bio-Helix®, Taiwan). Precision Plus Protein™ All Blue Prestained protein standard (Bio-Rad CA, USA) was used as a reference. Stained gels were documented using a Bio-Rad Gel Doc™ EZ and later analyzed using Image Lab or ImageJ software.
Mass culturing and purification of apoA-I∆43 mutant by affinity chromatography
We performed mass cultivation on 2-liter flasks. At a predetermined time after IPTG induction, whole culture media was collected and centrifuged to obtain the bacteria pellet. The pellet was stored at -80°C until extraction.
To begin the extraction, the cell pellet was dissolved in binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 40 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (PMSF), pH 7.4) at 10 mL per 1 g of cell pellet. Cell membrane disruption was achieved using JY88IN ultrasonic homogenizer (Drawell, Chongqing, China) at 4°C. Soluble protein fraction and insoluble protein pellet were separated by means of centrifugation. Immobilized Metal Ion Affinity Chromatography separation of polyhistidine-tagged recombinant products was performed using nickel-chelated resin (Ni-Sepharose™ 6 Fast Flow, Cytiva Life Sciences, USA) which was manually packed into Tricorn™ 10/100 empty chromatography column according to manufacturer recommendations. The void volume of this manually packed column was 7.5 mL. The column was preconditioned with a binding buffer at the flow rate of 5 mL/min. After stable UV280 baseline was achieved, the clarified soluble protein fraction was loaded into chromatographic column at 2.5 mL/min flow rate. After washing the unbound proteins from the column with binding buffer at 5 mL/min, tagged recombinant products were eluted from Ni-resin by gradient addition of elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, 1 mM PMSF, pH 7.4). Purification process and purified fragment collection was performed using ÄKTA™ start protein purification system (Cytiva Life Sciences, USA). Eluted recombinant products were dialyzed to remove imidazole and buffer replacement. Protein concentrations were determined using Bradford’s method. After freeze-dried, purified recombinant products were kept at -80°C until use.
Peptide sequencing by mass spectrometry
In-gel tryptic digestion
Purified recombinant products were separated using SDS-PAGE. Respective peptide bands on the gel were cut and analyzed. Each gel piece was destained by 50% acetonitrile (ACN) in 10 mM ammonium bicarbonate until colorless. Proteins were reduced by adding 4 mM DTT and incubated at 65°C for 30 min. The gel pieces were added 50 mM iodoacetamide (IAA) (Sigma-Aldrich, USA) and incubated at room temperature for 30 min in dark. The reaction was quenched by adding 4 mM DTT and incubated at room temperature for 5 min. All solutions were removed, and gel pieces were dehydrated by adding 100% ACN (Thermo Scientific, USA). The gels were dried at room temperature. Protein digestion was performed overnight at 37°C with trypsin (Thermo Scientific, USA) in 10 mM ammonium bicarbonate (Sigma-Aldrich, USA). The peptides were extracted by adding 100% ACN in each tube and incubated for 15 min. The supernatant was transferred into a new microcentrifuge tube. All supernatants were dried using centrifugal concentrator (TOMY, Japan). All samples were stored at −20 °C prior to mass spectrometric analysis.
Sample analysis
The samples were analyzed using Utimate3000 ultra-high-pressure system coupled to a Sciex TripleTOF 6600+ (Sciex, USA). The peptides were separated in reversed-phase mode using Pepmap100 C18 at a column temperature of 55°C. The dimensions of the columns were 75 μm internal diameter, 15 cm length and 1.9 μm particle size. Liquid chromatography (LC) conditions: mobile phase A (A) composed of 0.1% formic acid (FA) in water and mobile phase B (B) comprising 80% acetonitrile with 0.04% FA. The samples were loaded onto nano analytical column was first linear gradient separated according to 95 minutes from 3-35 % B from the nano-LC system at a constant flow rate of 300 nL/min. The analytical column was regenerated at 90% B for 10 minutes and re-equilibrated at 5% B for 15 minutes. Precursor masses were collected in the mass range of 400 to 1,500 m/z with 250 ms in “high sensitivity” mode. Further fragmentation of each precursor spectrum occurred with a maximum of 30 precursors per cycle. The raw MS-spectra files (.wiff) were analyzed with Peak Software.
Reconstitution of high-density lipoprotein nanoparticles
rHDL reconstitution was performed using the urea-assisted cholate dialysis method and purified by density gradient ultracentrifugation (DGU) (Jonas, 1986; Suda et al., 2017; Fukuda et al., 2020). Briefly, apoA-I∆43 mutant and DSPC were dissolved in 4M urea in PBS and 30 mg/mL sodium cholate in 0.9% NaCl, respectively. The solutions were mixed and allowed to react at DSPC phase-transitioning temperature (56°C) for 30 minutes. The molar ratio between apoA-I∆43 mutant and DSPC was kept at 1:200. Subsequently, the mixture was transferred into a dialysis bag (50 kDa, MWCO) and dialyzed against PBS at phospholipid phase transition temperature and gradually cooled down to room temperature. During the dialysis, urea and sodium cholate were removed to allow the formation of rHDL. After the removal of debris by centrifugation, the density of this rHDL crude mixture was adjusted to 1.31 g/mL using solid KBr. This mixture was then loaded into an ultracentrifuge tube (Ultra-Clear™ tubes, Beckman Coulter, CA, USA). Density gradient layers were constructed by the sequential addition of 1.21, 1.063 and 1.019 g/mL KBr in 0.9% NaCl solutions into the ultracentrifuge tubes. Ultracentrifugation was conducted at 286,000 g, 16°C for 2 hours in Backman Optima L-XP floor-standing ultracentrifuge. The purified rHDL samples were collected from fractions in the 1.21 to 1.063 g/mL density layer and concentrated using Vivaspin® 20 centrifugal concentrator (Sartorius Stedim Lab Ltd., Gloucestershire, UK). The concentrated rHDL solution was dialyzed overnight against PBS (50 kDa, MWCO), then filtered and stored at 4°C before use.
For the preparation of coumarin-6 tagged rHDL mutant (C6-HDL), DSPC and coumarin-6 at a molar ratio of 8:0.04 were dissolved in chloroform in a round-bottom flask. The flask was heated with water bath to 40°C and the thin lipid film was created using a rotary evaporator. Subsequently, the thin lipid film was rehydrated with sodium cholate and subsequently mixed with apoA-I∆43 using the same protocol as described earlier.
rHDL characterization
Dynamic light scattering (DLS)
DLS was used to measure rHDL hydrodynamic size. rHDL sample was loaded into disposable cuvette and analyzed by ZetaSizer® Nano (Malvern Panalytical, Worcestershire, United Kingdom). Size distribution by volume was used as method of choice for particle size calculations.
Non-denatured native-PAGE analysis
Native PAGE was used to study the structure and composition of rHDL in native, non-denatured conditions. 10 µL of rHDL samples were mixed with 10 µL of native-PAGE sample buffer 2X (6.25 mM Tris-HCl, 25% glycerol, 1% bromophenol blue) before being loaded into a precast 4-20% gradient gel (Mini-PROTEAN®TGX™, Bio-Rad, CA, USA). The separation was conducted in native-PAGE running buffer consisting of 25 mM Tris-HCl and 192 mM glycine (pH 8.6), at 150 V constant voltage and a maximum current limit of 40 mA. Precision Plus Protein™ All Blue Prestained protein standard (Bio-Rad, CA, USA) was used as reference. Gel documentation and molecular weight calculation were carried out using Bio-Rad Gel Doc™ EZ imager and Image Lab software.
High-performance liquid chromatography size exclusion chromatography (HPLC-SEC) analysis.
HPLC-SEC was used to evaluate the size distribution of rHDL. Filtered rHDL samples were injected into Shimadzu Prominence™ modular HPLC system equipped with LC-20AD pumps, SIL-20A autosampler, CTO-20A column oven, and SPD-20A UV-Vis detector. Separation was performed using an SEC column (TSKgel® G3000SWXL 7.8 × 300 mm., 5 µm particle size, Tosoh Biosciences, Tokyo, Japan). The mobile phase consisting of 150 mM NaCl, 50 mM Tris, and 2 mM EDTA (pH 7.4) was delivered isocratically at 1.0 mL/min. Precision Plus Protein™ All Blue Prestained protein standard (Bio-Rad CA, USA) was used as a reference. All SEC separations were monitored at UV 280 nm.
RESULTS
Optimal condition for recombinant expressing of apoA-IΔ43 by BL21 Star™ (DE3) chemically competent E. coli.
Effects of temperature, induction time and duration of recombinant product expression
We investigated apoA-IΔ43 expression levels under various expression conditions. Results are shown in Figure 2(A). Comparative SDS-PAGE showed that, after the induction of recombinant protein expression by adding IPTG (at a working concentration of 0.5 mM) to the culture medium, new, distinctive bands were observed. The observed molecular weight (30 kDa) is in accordance with the expected molecular weight of apoA-IΔ43. These findings suggested that the recombinant apoA-IΔ43 was successfully synthesized. Further examination revealed that, during IPTG induction at 37°C, a time-dependent increase in 30 kDa band intensity was observed from 1-3 hours, with the strongest band intensity at 3 hours after IPTG addition. However, at 6 hours after IPTG addition, the 30 kDa band intensity was weakened compared to that observed after 3 hours. The 30 kDa band almost disappeared after overnight induction. Meanwhile, decreasing the induction temperature to 28ºC did not improve the expression level of apoA-IΔ43. The intensities of all visible protein bands as well as the 30 kDa band, were weaker. The 30 kDa band completely disappeared after overnight induction at 28ºC. Therefore, we concluded that the optimal induction temperature and duration for apoA-IΔ43 expression using BL21 Star™ (DE3) chemically competent cells are 37°C for 3 hr.
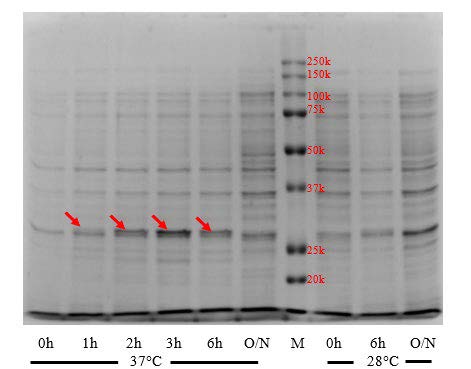
Figure 2(A). Effects of induction temperature and duration on apoA-IΔ43 expression level. Red arrows indicated 30kDa band. (M: marker, O/N: overnight)
Effects of IPTG concentration on recombinant product expression
We examined the effects of IPTG concentration on the expression level of apoA-IΔ43 after 3-hour induction at 37°C. IPTG concentrations ranging from 0.1 to 1.0 mM were evaluated. Results are shown in Figure 2(B). We observed the 30 kDa-bands at all induction concentrations. However, there was no significant difference between 30kDa-bands at each IPTG concentrations, suggesting that the minimum IPTG concentration was sufficient to initiate the expression of the recombinant product. Therefore, we selected the lowest concentration (0.1 mM) as the optimal IPTG concentration required induce the expression of apoA-IΔ43.
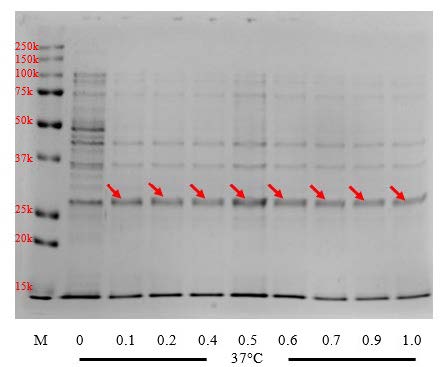
Figure 2(B). Effects of IPTG concentration on apoA-IΔ43 expression level. Red arrows indicated the 30kDa band. (M: marker, 0 – 1.0: IPTG concentration in millimolar)
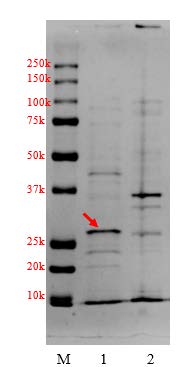
Figure 2(C). Localization of apoA-IΔ43 in harvested crude cell pellet and supernatant. (M: marker, 1: soluble fraction, 2: insoluble pellet)
Extraction and purification of recombinant apoA-IΔ43 mutant using immobilized metal ion affinity chromatography
First, we investigated whether the recombinant products existed in the insoluble fraction (inclusion bodies) or in the soluble fraction of cell lysate. We separated the insoluble fraction by dissolving the harvested cells pellet in lysis buffer, followed by 3 freeze-thaw cycles in liquid nitrogen. The lysed cell suspension was centrifuged to separate the insoluble fraction pellet from the soluble fraction supernatant. Results are shown in Figure 2(C). The strong band at the 30-kDa region can be clearly observed in the soluble fraction (lane 1), suggesting that most of apoA-IΔ43 had existed in the soluble form.
In the production run, we collected and clarified the soluble fraction using 2.5 µm grade 5 qualitative filter paper (Whatman®, Cytiva Life Sciences, USA) followed by high-speed centrifugation. The clear protein solution was loaded into the FPLC system equipped with a column containing a nickel-chelated resin in a non-denatured form. Protein elution was observed by measuring UV absorbance at 280 nm (UV280). First, the clarified sample was loaded at a flow rate of 1 mL/min. However, we observed a significant loss of 30-kDa protein in the flow-through (Figure S1, lane 6-7). This suggested that there was apoA-IΔ43 leakage in the flow-through, which might have been due to suboptimal binding conditions. In the next purification cycle, the sample loading process was optimized by maintaining a flow rate of 2.5 mL/min. We also decreased the viscosity of the clarified supernatant by twofold dilution with the binding buffer prior to column loading. After sample loading, the washing was carried out using the binding buffer with at a flow rate of 5 mL/min for approximately 150 mL to ensure that all non-tagged proteins were completely eliminated. The results of this optimization are summarized in Figure 3. We observed that near the end of sample loading (Figure 3(A), lane 8), the 30-kDa band was significantly weaker than that observed in the unoptimized chromatographic condition even at the early phase of sample loading (Figure S1, lanes 6-7). There was no visible band in the eluted sample during the early phase of non-tagged protein washing (Figure 3(A), lane 9). These findings suggested that the binding of apoA-IΔ43 to the chromatographic resin was improved. To elute the his-tagged protein, the elution buffer was introduced into the mobile phase. We found that at a 1:1 ratio (binding: elution buffer), all his-tagged protein has been completely eluted from the column. As shown in Figure 3(A) lanes 4-6, strong 30 kDa bands were observed. Surprisingly, these bands were more intense than those observed in the pre-optimized chromatographic conditions (Figure S1, lane 1). The relative band intensities of 30-kDa bands found in the optimized chromatographic condition were stronger than those observed in the unoptimized chromatographic condition. These findings suggested that the optimized chromatographic condition had greatly improved apoA-IΔ43 recovery. The estimated recovery of apoA-IΔ43 using the optimized chromatographic condition was found to be approximately 20 mg per 1 L of culture volume and the purity of the extracted apoA-IΔ43 fractions were greater than 80%.
Mass-spectrometry protein sequencing of apoA-IΔ43
To verify the amino acid sequence of apoA-IΔ43, we performed SDS-PAGE separation on a diluted fraction of purified apoA-IΔ43. The 30-kDa bands were excised and processed with in-gel tryptic digestion before analyzing by LC-MS/MS. The results, shown in Figure 1, were compared to the 265-amino-acid reference sequence of apoA-IΔ43, which includes the terminal amino acids encoded by the pET100/D-topo vector. The LC-MS/MS analysis revealed substantial sequence coverage, with identified peptide fragments spanning 185 amino acids out of the total 265. These findings confirmed successful expression and verified the identity of the apoA-IΔ43 protein.
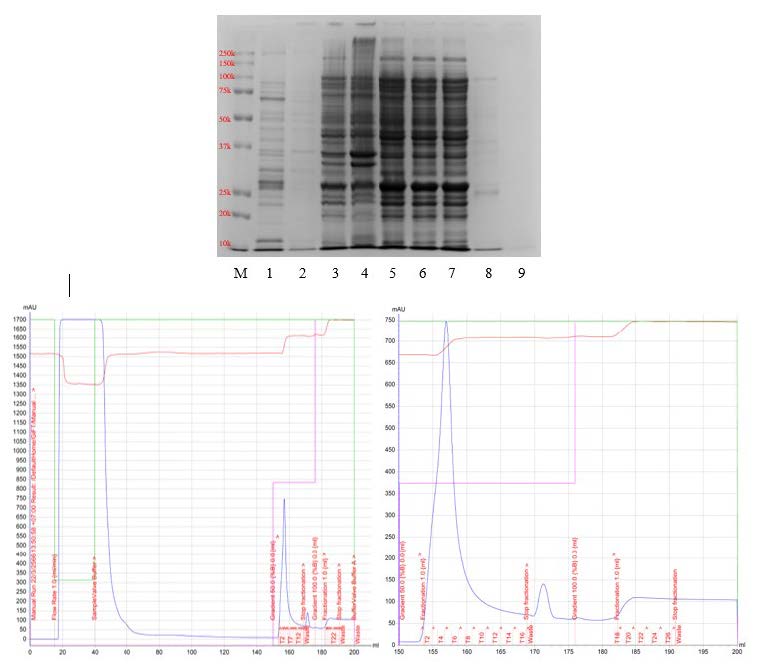
Figure S1. SDA-PAGE and FPLC chromatogram obtained from the initial purification of apoA-IΔ43 (lane definitions; M: marker, 1: pooled T3-T7, 2: pooled T1, T2 and T8, 3: crude cell lysate, 4: insoluble pellet, 5: soluble supernatant, 6-9: eluted sample at 30, 38, 55 and 140 mL). The chromatogram on the left shows a summary of the process, whereas the chromatogram on the right shows the details of the collected fractions.
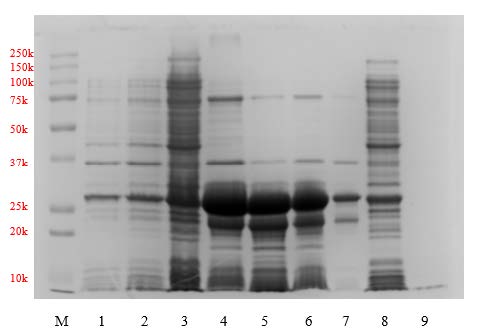
Figure 3(A). SDS-PAGE obtained during the purification of apoA-IΔ43 using the optimized loading procedure (lane definitions; M: marker, 1: crude cell lysate, 2: insoluble pellet, 3: soluble supernatant, 4: T4, 5: T7, 6: pooled T3-T10, 7: pooled T2, T11-13, 8-9: eluted sample at 95 and 125 mL, respectively.
Reconstitution and characterization of C6-labeled lipoprotein nanoparticles
Freeze-dried apoA-IΔ43 was rehydrated with 4M urea in PBS and was mixed with C6-labeled DSPC in sodium cholate at the phase transition temperature (56°C). After 30 minutes, both urea and sodium cholate were removed by dialysis, and the mixture was further purified by DGU. The unpurified nanoparticle solution was mixed with KBr to adjust its density to 1.31 g/mL and was loaded into an ultracentrifugation tube. After that, 4 density layers (1.21, 1.063, 1.019, and 1 g/mL) of KBr in PBS were constructed above the unpurified layer (Figure 4). By observing the position of the greenish layer of C6, we were able to determine the density of the reconstituted nanoparticle. As seen in Figure 4, after DGU, a strong green layer of C6 was observed in the 1.063 g/mL density layer, suggesting that the density of C6-labeled lipoprotein nanoparticle was consistent with natural HDL (1.21 to 1.063 g/mL). Hydrodynamic size analysis found that the C6-labeled lipoprotein nanoparticles had an average diameter of 11.7 ± 3.4 nm (Figure S2). Bradford analysis confirmed the presence of protein (0.914 mg/mL) in the sample. These findings suggested that both C6-labeled DSPC and apoA-IΔ43 were successfully integrated into the lipoprotein nanoparticles. In Figure 5, non-denatured native-PAGE analysis results showed a single band at 180 kDa and the SEC-HPLC chromatogram also showed a single peak at 7.17 minutes. There was no precipitation observed in the purified lipoprotein fractions stored at 4°C for 3 months.
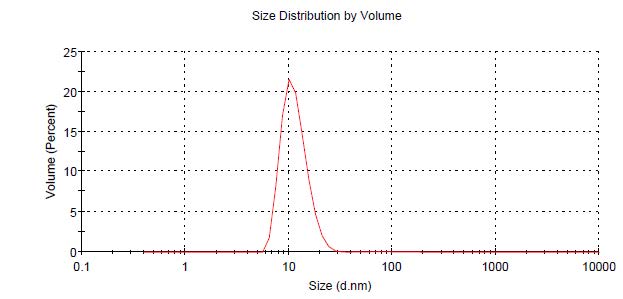
Figure S2. Hydrodynamic size distribution of C6-labelled reconstituted lipoprotein nanoparticles determined by Dynamic Light Scattering (DLS) method.
DISCUSSION
Recombinant protein expression technique allows us to produce the protein of interest and use it to study bioactivities, functions, or additional applications without supply constraint or ethical concern. We chose E. coli as the expression host because of its rapid doubling time, easy to maintain, has a low cost of production, and offers availability of compatible expression vectors with affinity tags (Balbas and Lorence, 2004; Rosano and Ceccarelli, 2014). Various pET vector series with affinity tags such as poly-histidine are also readily available. Its recombinant products can be easily extracted and purify using nickel-chelated affinity chromatography (Balbas and Lorence, 2004; Rosano and Ceccarelli, 2014; Ikon et al., 2017). Previously, we used E. coli as the expression host for the synthesis of GFP-tagged α-1,3-glucanase from Streptomyces thermodiastaticus HF3-3 (Panti et al., 2021). In this study, we adopted the N-terminal 43 amino acids deleted human apoA-I (apoA-IΔ43) sequence, which was reported to associate exclusively with plasma HDL and similar free energies of binding to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) vesicles (Borhani et al., 1997). In this studies, we developed a complete protocol for apoA-IΔ43 synthesis. In all conditions, we initiated IPTG induction at the mid-log phase (OD600 ~ 0.6). First, we evaluated whether the temperature during induction with IPTG would increase the expression of apoA-IΔ43. While the lac operator showed the highest activity at 37°C, the use of a suboptimal temperature might help decrease the unwanted metabolic responses to the synthesis of foreign proteins and improve the production, protein folding, and/or solubility of the target protein product (Chalmers et al., 1990; Donovan et al., 1996). The soluble expression is the result of a properly folded recombinant product, and it should be biologically active without the need for further refolding (Rosano and Ceccarelli, 2014; Kasekarn et al., 2020). Our findings in Figure 2(A) showed that at 37°C, the expression level of apoA-IΔ43 was significantly higher than at 28°C. A further reduction of the induction temperature to 20°C resulted in the complete abolition of apoA-IΔ43 production (data not shown). We also found that at 37°C, most of the apoA-IΔ43 already existed in a soluble form (Figure 2(C)). Since we were required to denature apoA-IΔ43 during the urea-assisted cholate-dialysis reconstitution method (Fukuda et al., 2020), we prioritized the expression yield and concluded that 37°C was our optimal induction temperature.
The duration of the IPTG induction period also plays a significant role in recombinant protein expression. As shown in Figure 2(A), the apoA-IΔ43 expression level increased as the IPTG induction progresses until reaching the three-hour point. Subsequently, the expression level diminished and almost completely disappeared after overnight induction. This decline can likely be attributed to the metabolic burden on the bacterial host, protein degradation by intracellular proteases, formation of inclusion bodies, or cellular toxicity resulting from prolonged exposure to IPTG and overexpression stress (Gopal and Kumar, 2013; Rosano and Ceccarelli, 2014).
After that, we investigate at which IPTG concentration yielded the highest expression of apoA-IΔ43. In Figure 2(B), we observed that an IPTG concentration as low as 0.1 mM sufficiently initiated apoA-IΔ43 production. Despite the manufacturer’s expression guidelines recommending a higher IPTG concentration (0.5 mM), we did not observe any increase in the apoA-IΔ43 expression level as the IPTG concentration level increased beyond 0.1 mM. This might be because IPTG at 0.1 mM was likely sufficient to saturate the lac repressor protein (lacI) or achieve the saturation of T7 RNA polymerase (Balbas and Lorence, 2004; Rosano and Ceccarelli, 2014). Research indicates that lacI inhibition and subsequent full induction of the lac operon can be achieved at relatively low IPTG levels. For example, studies have demonstrated that induction begins at concentrations as low as 8–10 µM, with maximal activity often observed above 40 µM in some systems (Fernández-Castané et al., 2012; Tan et al., 2012). In addition, E. coli cells uptake IPTG rapidly through a lacY-mediated active transport. This saturable process is a rate-limiting step in the induction of recombinant protein expression (Simas et al., 2023). Additionally, since the passive diffusion of IPTG across the membrane was limited (Simas et al., 2023), increasing the IPTG concentration beyond the saturated level is unlikely to increase the expression level of the recombinant product.
Extraction and purification of apoA-IΔ43 followed the basic principles of affinity chromatography. The pET100/D-topo vector encodes six histidine residues upstream of the sequence apoA-IΔ43. These histidine residues interact with nickel-chelated resin primarily through coordination bonding between the imidazole nitrogen atoms of histidine and immobilized nickel ions (Ni2⁺) on the resin matrix. This specific and reversible binding allows effective purification from complex protein mixtures. The imidazole group of histidine coordinates directly with the nickel ion via nitrogen atoms, forming stable complexes. Upon binding, proteins can be selectively eluted by competition with free imidazole molecules or by altering the pH, thus disrupting the metal-histidine coordination (Block et al., 2009). In our initial findings in Figure S1, significant loss of the recombinant product was observed due to poor binding conditions. According to the manual, the binding capacity of the affinity column that we used was up to 200 mg of His-tagged protein. Our obtained tagged protein concentration was low, and the total amount was much lower than the capacity of the column (<50 mg/batch). Therefore, we believed that the tagged protein leakage might have been due to the weak affinity tag interaction between the folded tagged protein and the nickel-chelated resin (Block et al., 2009). However, later examination with denatured cell lysate did not increase the binding (data not shown). Therefore, to improve the retention of tagged protein, we increase the column residence time by reducing the flow rate during cell lysate loading to half of the recommended rate (2.5 mL/min). Extending the residence time enhances the interaction between immobilized metal ions and His-tagged proteins, thereby facilitating stronger binding and improved adsorption efficiency. This is particularly beneficial when the target protein concentration is low or when contaminants compete for binding sites (Block et al., 2009). The results in Figure 3(A) showed significant improvements in protein retention. However, a notable increase in non-tagged protein retention was also observed. This increase in nonspecific binding occurs because prolonging column residence time at lower flow rates allows more opportunities for weak, nonspecific interactions between impurities and the immobilized metal ions or resin matrix, thus decreasing overall selectivity (Bornhorst and Falke, 2000; Bolanos-Garcia and Davies, 2006; Block et al., 2009). Therefore, we believed that more intensive washing and/or pre-elution processes were required.
In Figure 1, the protein sequence coverage obtained from LC-MS/MS analysis provides critical insights into protein identification and structural characterization. The human apoA-I gene has two exons, the second exon encodes amino acids numbers 44 to 243 (Fukuda et al., 2020). In the current study, a significant portion of apoA-IΔ43 was confidently identified through peptide mapping, yielding comprehensive sequence coverage across multiple distinct regions of the protein. We found that most of the missing regions were N-terminal vector-derived extensions which do not impact the protein function. This extensive coverage underscores the effectiveness of the employed proteomic strategy, including the enzymatic digestion protocol and analytical conditions optimized for peptide detection by LC-MS/MS (Zhang et al., 2013; Aebersold and Mann, 2016). Highlighted peptide sequences identified span throughout the protein's length, indicating successful enzymatic digestion, primarily by trypsin, known for cleaving at the carboxyl side of lysine (K) and arginine (R) residues (Olsen et al., 2004). The identified peptides, including regions such as "DNWDSVTSTF," "SKLREQLGPV," and "LEALKENGGA," suggest robust ionization and detection efficiencies. Conversely, gaps or regions of poor coverage observed in the protein sequence could reflect factors such as incomplete proteolysis, low peptide solubility, poor ionization efficiency, or potential post-translational modifications that alter the peptide masses beyond the anticipated range (Steen and Mann, 2004; Tsiatsiani and Heck, 2015).
To verify the activities of apoA-IΔ43, a simple HDL reconstitution reaction using the purified apoA-IΔ43 and phospholipid DSPC was examined. ApoA-IΔ43, an N-terminal 43-amino acid truncated apoA-I mutant, was reported to be a protein component of a discoidal HDL that has been characterized in a native-like environment, and discoidal HDL prepared with this truncated apoA-I was reported to have similar cellular cholesterol efflux capabilities compared to those of wild-type HDL (Davidson et al., 1995; Borhani et al., 1997; Gursky et al., 2012; Fukuda et al., 2020). Using this N-terminal truncated apoA-I variant should improve expression efficiency while maintaining minimal impact on its bioactivities. Furthermore, N-terminal deletion creates open space for protein fusion, which can be fused to the existing amino acids 44-243 backbone. To prepare synthetic HDL, synthetic phosphatidylcholine such as DSPC was used. When apoA-I is mixed with phospholipids, spontaneous self-assembly occurs to form discoidal HDL-like particles (Jonas, 1986). The insertion of apoA-I into lipid the membrane is the rate-limiting step of this reaction, and urea was used to increase the reaction rate and improve the reconstitution yield (Fukuda et al., 2020). During apoA-I insertion, apoA-I induces significant reorganization of phospholipid arrangement. Rather than existing in extended bilayer configurations, phospholipids become arranged in more compact, curved structures around the apoA-I scaffold (Jonas, 1986; Gursky et al., 2012; Fukuda et al., 2020). This leads to an increase in the density of HDL-like nanoparticles compared to apoA-I-absent phospholipid nanoparticles. Based on this density change, the successful formation of rHDL can be determined by DGU. To achieve clear separation and identification, we employed potassium bromide (KBr) DGU, a well-established technique for isolating HDL nanoparticles from other components based on distinct density differences (Chapman et al., 1981). Following ultracentrifugation, a distinct green layer corresponding to C6-labeled nanoparticles was clearly visible within the density gradient of 1.21-1.063 g/mL (Figure 4). This specific density range aligns closely with that reported for natural human HDL, indicating successful reconstitution of HDL-like nanoparticles (Chapman et al., 1981). To further validate the presence and purity of HDL nanoparticles, Bradford’s protein analysis was performed on the isolated fractions. Protein was detected exclusively in samples collected from the HDL-associated density layers (1.063–1.21 g/mL), confirming the protein-lipid composition characteristic of natural HDL (Suda et al., 2017; Fukuda et al., 2020). In contrast, protein was absent in fractions outside this specific density range, underscoring the selectivity and efficacy of our isolation process. Non-denatured PAGE analysis showed that C6-labeled nanoparticles have a molecular weight of approximately 180 kDa (Figure 5). The molecular weight of native apoA-I (~28 kDa) typically leads to rHDL particles with sizes ranging between 150 and 300 kDa, depending upon lipid composition and protein-to-lipid ratios (Jonas 1986; Suda et al. 2017; Fukuda et al., 2020). In our formulation, apoA-IΔ43 has an estimated molecular weight of approximately 30 kDa, suggesting that multiple apoA-IΔ43 copies are associated with each DSPC micelle. The measured 180 kDa rHDL particle would indicate incorporation of 3-4 copies of apoA-IΔ43 within each rHDL nanoparticle, with the remaining mass (~90–110 kDa) contributed primarily by the lipid component. This integration of 3-4 copies of apoA-I into a single rHDL particle was similarly reported in previous studies (Sparks et al., 1992; Cedó et al., 2016; Fukuda et al., 2023). These findings suggest that our purified apoA-IΔ43 is biologically active and suitable for use in further rHDL biological studies, drug delivery vehicles, and other pharmaceutical applications.

Figure 4. Lipoprotein nanoparticles sample before (A) and after (B) density gradient ultracentrifugation.
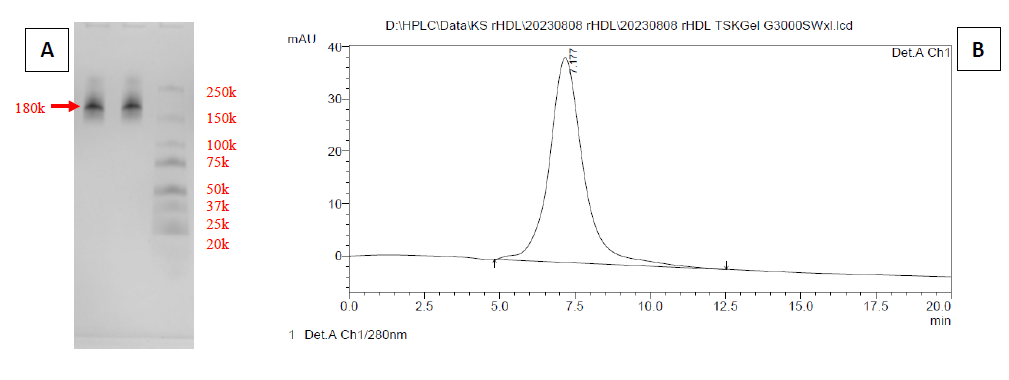
Figure 5. Size-exclusion analysis of lipoprotein nanoparticles by native-PAGE (A) and SEC-HPLC chromatogram (B).
CONCLUSIONS
In this study, we developed a complete upstream/downstream process for in-house recombinant synthesis of apoA-IΔ43 for use in rHDL application studies. We were able to efficiently synthesize apoA-IΔ43 with sufficient purity. The obtained apoA-IΔ43 was sequence-confirmed. The biological activities were also evaluated and confirmed. We are looking forward to examining the further applications of rHDL as the drug delivery platform and its anti-inflammatory activities. To our knowledge, this report is the first successful rHDL reconstitution using apoA-IΔ43 and DSPC in Thailand.
ACKNOWLEDGMENTS
The authors would like to express our gratitude to Faculty of Pharmacy, Faculty of Agro-Industry, Chiang Mai University for providing instruments.
AUTHOR CONTRIBUTIONS
Niphawan Panti performed experiments, data analysis and wrote a manuscript. Sasithorn Sirilun provided facilities and crucial guidance for the project. Jakkaphan Kumsab and Yodying Yingchutrakul performed LC/MS analysis and interpretation of the data. Kasirawat Sawangrat designed, performed experiments, data analysis and wrote the manuscript. All authors have read and approved of the final manuscript.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
AbdelHafez, M.A. 2022. Protective and therapeutic potentials of HDL and ApoA1 in COVID-19 elderly and chronic illness patients. Bulletin of the National Research Centre. 46(1): 222.
Aebersold, R. and Mann, M. 2016. Mass-spectrometric exploration of proteome structure and function. Nature. 537(7620): 347–355.
Balbas, P. and Lorence, A. 2004. Recombinant gene expression. New Jersey: Humana Press.
Ben-Aicha, S., Badimon, L., and Vilahur, G. 2020. Advances in HDL: Much more than lipid transporters. International Journal of Molecular Sciences. 21(3): 732.
Block, H., Maertens, B., Spriestersbach, A., Brinker, N., Kubicek, J., Fabis, R., Labahn, J., and Schäfer, F. 2009. Immobilized-metal affinity chromatography (IMAC): A review. Methods in Enzymology. 463: 439–473.
Bolanos-Garcia, V.M. and Davies, O.R. 2006. Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. Biochimica et Biophysica Acta (BBA) - General Subjects. 1760(9): 1304–1313.
Borhani, D.W., Rogers, D.P., Engler, J.A., and Brouillette, C.G. 1997. Crystal structure of truncated human apolipoprotein A-I suggests a lipid bound conformation. Proceedings of the National Academy of Sciences. 94(23): 12291–12296.
Bornhorst, J.A. and Falke, J.J. 2000. Purification of proteins using polyhistidine affinity tags. Methods in Enzymology. 326: 245–254.
Cedó, L., García-León, A., Baila-Rueda, L., Santos, D., Grijalva, V., Martínez-Cignoni, M.R., Carbó, J.M., Metso, J., López-Vilaró, L., Zorzano, A., et al. 2016. ApoA-I mimetic administration, but not increased apoA-I-containing HDL, inhibits tumour growth in a mouse model of inherited breast cancer. Scientific Reports. 6(1): 36387.
Chalmers, J.J., Kim, E., Telford, J.N., Wong, E.Y., Tacon, W.C., Shuler, M.L., and Wilson, D.B. 1990. Effects of temperature on Escherichia coli overproducing beta-lactamase or human epidermal growth factor. Applied and Environmental Microbiology. 56(1): 104–111.
Chapman, M.J., Goldstein, S., Lagrange, D., and Laplaud, P.M. 1981. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. Journal of Lipid Research. 22(2): 339–358.
Cochran, B.J., Ong, K.L., Manandhar, B., and Rye, K.A. 2021. APOA1: A protein with multiple therapeutic functions. Current Atherosclerosis Reports. 23(3): 11.
Davidson, W.S., Gillotte, K.L., Lund-Katz, S., Johnson, W.J., Rothblat, G.H., and Phillips, M.C. 1995. The effect of high density lipoprotein phospholipid acyl chain composition on the efflux of cellular free cholesterol. The Journal of Biological Chemistry. 270(11): 5882–5890.
Deuringer, B., Härdtner, C., Krebs, K., Thomann, R., Holzer, M., Hilgendorf, I., and Süss, R. 2022. Everolimus-loaded reconstituted high-density lipoprotein prepared by a novel dual centrifugation approach for anti-atherosclerotic therapy. International Journal of Nanomedicine. 17: 5081–5097.
Donovan, R.S., Robinson, C.W., and Glick, B.R. 1996. Review: Optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter. Journal of Industrial Microbiology. 16(3): 145–154.
Fernández-Castané, A., Caminal, G., and López-Santín, J. 2012. Direct measurements of IPTG enable analysis of the induction behavior of E. coli in high cell density cultures. Microbial Cell Factories. 11: 58.
Fox, C.A., Moschetti, A., and Ryan, R.O. 2021. Reconstituted HDL as a therapeutic delivery device. Biochimica et Biophysica Acta. Molecular and Cell Biology of Lipids. 1866(11): 159025.
Fukuda, R., Mahmuda, N., Kasirawat, S., Kawakami, R., Shima, R., Mizukami, Y., Shibukawa, S., Tada, Y., Kawanishi, F., Ogura, M., et al. 2023. High density lipoprotein engineering for eye drop treatment of age-related macular degeneration. Advanced Therapeutics. 6(11): 2300186.
Fukuda, R., Saito, M., Shibukawa, S., Sumino, A., Nakano, M., and Murakami, T. 2020. Urea-assisted reconstitution of discoidal high-density lipoprotein. Biochemistry. 59(15): 1455–1464.
Fukuda, R., Tani, M., Shibukawa, S., Nobeyama, T., Nomura, T., Kato, Y., and Murakami, T. 2023. Effects of lipoprotein nanoparticles’ composition and size on their internalization in plant and mammalian cells. Genes to Cells. 28(12): 881–892.
Gopal, G.J. and Kumar, A. 2013. Strategies for the production of recombinant protein in Escherichia coli. The Protein Journal. 32(6): 419–425.
Gordon, S.M., Hofmann, S., Askew, D.S., and Davidson, W.S. 2011. High density lipoprotein: It's not just about lipid transport anymore. Trends in Endocrinology and Metabolism. 22(1): 9–15.
Gursky, O., Mei, X., and Atkinson, D. 2012. The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry. 51(1): 10–18.
Havel, R.J., Eder, H.A., and Bragdon, J.H. 1955. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. The Journal of Clinical Investigation. 34(9): 1345–1353.
Ikon, N., Shearer, J., Liu, J., Tran, J.J., Feng, S., Kamei, A., Beckstead, J.A., Kiss, R.S., Weers, P.M., Ren, G., et al. 2017. A facile method for isolation of recombinant human apolipoprotein A-I from E. coli. Protein Expression and Purification. 134: 18–24.
Jonas, A. 1986. Reconstitution of high-density lipoproteins. Methods in Enzymology. 128: 553–582.
Kasekarn, W., Suksiriphattanapong, B., Chokepaichitkool, T., Wanachewin, O., Roytrakul, S., and Kongtawelert, P. 2020. Soluble expression and purification of bioactive recombinant human bone morphogenetic protein-2 from Escherichia coli. Chiang Mai University Journal of Natural Sciences. 19(4): 752-773.
Lerch, P.G., Förtsch, V., Hodler, G., and Bolli, R. 1996. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sanguinis. 71(3): 155–164.
Navab, M., Anantharamaiah, G.M., Reddy, S.T., Van Lenten, B.J., Datta, G., Garber, D., and Fogelman, A.M. 2004. Human apolipoprotein A-I and A-I mimetic peptides: Potential for atherosclerosis reversal. Current Opinion in Lipidology. 15(6): 645–649.
Olsen, J.V., Ong, S-E., and Mann, M. 2004. Trypsin cleaves exclusively c-terminal to arginine and lysine residues. Molecular & Cellular Proteomics. 3(6): 608–614.
Panti, N., Cherdvorapong, V., Itoh, T., Hibi, T., Suyotha, W., Yano, S., and Wakayama, M. 2021. Functional analysis of α-1,3-glucanase domain structure from Streptomyces thermodiastaticus HF3-3. The Journal of General and Applied Microbiology. 67(3): 85–91.
Rogers, D.P., Brouillette, C.G., Engler, J.A., Tendian, S.W., Roberts, L., Mishra, V.K., Anantharamaiah, G.M., Lund-Katz, S., Phillips, M.C., and Ray, M.J. 1997. Truncation of the amino terminus of human apolipoprotein A-I substantially alters only the lipid-free conformation. Biochemistry. 36(2): 288–300.
Rosano, G.L. and Ceccarelli, E.A. 2014. Recombinant protein expression in Escherichia coli: Advances and challenges. Frontiers in Microbiology. 5: 172.
Ryan R.O. 2008. Nanodisks: Hydrophobic drug delivery vehicles. Expert Opinion on Drug Delivery. 5(3): 343–351.
Simas, R.G., Pessoa Junior, A., and Long, P.F. 2023. Mechanistic aspects of IPTG (isopropylthio-β-galactoside) transport across the cytoplasmic membrane of Escherichia coli-a rate limiting step in the induction of recombinant protein expression. Journal of Industrial Microbiology & Biotechnology. 50(1): kuad034.
Sparks, D.L., Lund-Katz, S., and Phillips, M.C. 1992. The charge and structural stability of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. Journal of Biological Chemistry. 267(36): 25839–25847.
Steen, H. and Mann, M. 2004. The ABC's (and XYZ's) of peptide sequencing. Nature Reviews. Molecular Cell Biology. 5(9): 699–711.
Suda, K., Murakami, T., Gotoh, N., Fukuda, R., Hashida, Y., Hashida, M., Tsujikawa, A., and Yoshimura, N. 2017. High-density lipoprotein mutant eye drops for the treatment of posterior eye diseases. Journal of Controlled Release. 266: 301–309.
Tan, J.S., Ramanan, R.N., Ling, T.C., Mustafa, S., and Ariff, A.B. 2012. The role of lac operon and lac repressor in the induction using lactose for the expression of periplasmic human interferon-α2b by Escherichia coli. Annals of Microbiology. 62(4): 1427–1435.
Tsiatsiani, L. and Heck, A.J. 2015. Proteomics beyond trypsin. The FEBS Journal. 282(14): 2612–2626.
van der Vorst EPC. 2020. High-density lipoproteins and apolipoprotein A1. p. 399-420. In: Hoeger, U., Harris, J. [ed] Vertebrate and invertebrate respiratory proteins, lipoproteins and other body fluid proteins. Subcellular Biochemistry. Vol. 94. Cham: Springer.
Zhang, W., Wang, J., Jia, J., Chen, L., Wu, Z., and Liu, J. 2015. A simple method to extract whole apolipoproteins for the preparation of discoidal recombined high density lipoproteins as bionic nanocarriers for drug delivery. Journal of Pharmacy & Pharmaceutical Sciences. 18(2): 184–198.
Zhang, Y., Fonslow, B.R., Shan, B., Baek, M-C., and Yates, J.R. 2013. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113(4): 2343–2394.
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand. https://cmuj.cmu.ac.th
Niphawan Panti1, Sasithorn Sirilun2, 3, Jakkaphan Kumsab4, Yodying Yingchutrakul4, and Kasirawat Sawangrat2, *
1 Division of Biotechnology, Faculty of Agro-Industry, Chiang Mai University, Chiang Mai 50100, Thailand.
2 Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand.
3 Innovation Center for Holistic Health, Nutraceuticals, and Cosmeceuticals, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand.
4 National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Pathum Thani 12120, Thailand.
Corresponding author: Kasirawat Sawangrat, E-mail: kasirawat.s@cmu.ac.th
ORCID: Kasirawat Sawangrat: https://orcid.org/0000-0002-0936-1588
Total Article Views
Editor: Sirasit Srinuanpan,
Chiang Mai University, Thailand
Article history:
Received: July 8, 2025;
Revised: August 25, 2025;
Accepted: August 26, 2025;
Online First: September 19, 2025