 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

First Case of Compound Heterozygotes for Hb G-Georgia (HBA2:c.287C>T)/α0-thalassemia --SEA Deletion Found in Northern Thailand
Moe Theingi, Chedtapak Ruengdit, Manoo Punyamung, and Sakorn Pornpraset*Published Date : August 20, 2025
DOI : https://doi.org/10.12982/NLSC.2025.068
Journal Issues : Number 4, October-December 2025
Abstract Hemoglobin (Hb) G-Georgia is an α-globin variant caused by a mutation at codon 95 which substitutes the amino acid proline with a leucine. Heterozygosity for Hb G-Georgia may cause mild anemia, even though it is often asymptomatic. The Hb G-Georgia homozygosity or compound heterozygosity with other thalassemias can result microcytic hypochromic anemia with mild splenomegaly. We report the first case of compound heterozygosity for Hb G-Georgia/α0-thalassemia --SEA deletion (αGGα/--SEA) in a 62-year-old Thai woman living in Chiang Mai, northern Thailand. She presented with clinical symptoms of fatigue and dyspnea. Her hematological features revealed microcytic hypochromic anemia, but she had never received a blood transfusion. Hb analysis using capillary electrophoresis (CE) showed the peak of Hb G-Georgia (50.8%) in zone 7 (HbF zone). High-performance-liquid-chromatography (HPLC) showed coelution with HbA at the retention time of 2.36 minutes, and premier-resolution HPLC (PR-HPLC) detected 52.2% Hb G-Georgia but without clear separation from HbA. Next-generation sequencing confirmed the HBA2:c.287C>T mutation, and a single-tube multiplex real-time PCR with EvaGreen and high-resolution melting (HRM) analysis detected α0-thalassemia --SEA type deletion. Therefore, understanding genetic interactions between this α-globin variant and α0-thalassemia can help a better disease management and appropriate genetic counseling.
Keywords: Detection, α0-thalassemia, Hb G-Georgia, High performance liquid chromatography, Northern Thailand
Funding: MT had been supported by the CMU Presidential Scholarship from Chiang Mai University, Chiang Mai, Thailand since academic year 2024. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Citation: Theingi, M., Ruengdit, C., Punyamung, M., and Pornpraset, S. 2025. First case of compound heterozygotes for Hb G-Georgia (HBA2:c.287C>T)/α0-thalassemia --SEA deletion found in northern Thailand. Natural and Life Sciences Communications. 24(4): e2025068.
INTRODUCTION
Hemoglobin (Hb) variants, or abnormal Hb, are hemoglobinopathies resulting from mutations in the α-and β-globin genes. These mutations can lead to changes in the structure and function of Hb, which can result in various clinical manifestations, from asymptomatic carriers to severe hemolytic anemia (Tepakhan et al., 2024). One of the Hb variants is Hb G-Georgia [α95 (G2) Pro → Leu (α2) (HBA2:c.287C>T)], which is caused by a point mutation in the α-globin gene (HBA2). The mutation at codon 95 of the α2-globin gene causes the substitution of proline with leucine. Heterozygosity for Hb G-Georgia is often asymptomatic, with Hb G-Georgia levels generally below 25% (Huisman et al., 1970). However, homozygosity for Hb G-Georgia or compound heterozygosity with other thalassemia mutations can lead to microcytic hypochromic anemia and may sometimes present with mild splenomegaly. In certain populations, particularly in regions with high rates of hemoglobinopathies such as Southeast Asia, Hb G-Georgia may coexist with other genetic abnormalities, making diagnosis and clinical features more complex (Fucharoen and Winichagoon, 1987). Routine diagnostic tools such as high-performance liquid chromatography (HPLC) and capillary electrophoresis (CE) are useful for detecting the Hb variants. However, due to the limitation in sensitivity in these techniques, next-generation sequencing (NGS) has become applicable for identification and characterization of complex or novel hemoglobinopathies (He et al., 2017). Combining HPLC and CE for Hb analysis can help in initial recognition and presumptive diagnosis of cases, followed by appropriate DNA analysis for confirmation (Srivorakun et al., 2014).
CASE PRESENTATION
In this study, the authors report about a 62-year-old Thai woman living in Chiang Mai, a province in northern Thailand, who presented with fatigue and dyspnea. Upon examination, she appeared pale and did not have hepatosplenomegaly. Her history revealed that she had never received a blood transfusion. A complete blood count (CBC) was analyzed by using an automated blood counter (Sysmex KX-21, Sysmex Corporation, Kobe, Japan). Laboratory findings are as follows: RBC 4.9 x 1012 cells/L, total Hb 99 g/L, PCV 0.32 L/L, MCV 64.4 fL, MCH 20.0 pg, MCHC 311 g/L, and RDW 17.6%. Due to the low levels of total Hb, MCV, and MCH, combined with a high level of RDW, she was diagnosed with microcytic hypochromic anemia. However, iron deficiency anemia could be excluded because her serum iron (SI = 49 mg/dL), total iron-binding capacity (TIBC = 223 mg/dL), transferrin saturation (TS = 22%), and serum ferritin (SF = 197 mg/L) were observed within normal values. Therefore, her blood sample was sent to the Associated Medical Sciences-Clinical Service Center (AMS-CSC), Chiang Mai University, Chiang Mai, Thailand, for thalassemia diagnosis. In the thalassemia laboratory of the AMS-CSC, hemoglobin analysis was initially performed using the CE (Capillarys 2 Flex piercing, Sebia, Evry, France). The peak of Hb G-Georgia (50.8%) was found in zone 7 (HbF zone) of the CE electropherogram (Figure 1A). Then, HPLC (VARIANT β-thalassemia Short Program; Bio-Rad Laboratories, Hercules, CA, USA) was performed. The results showed that the Hb G-Georgia was coeluted with HbA at the retention time of 2.36 minutes on HPLC chromatogram (Figure 1B). In addition, premier resolution-high-performance liquid chromatography (PR-HPLC; Trinity Biotech, Co. Wicklow, Ireland) detected 52.2% Hb G-Georgia but without clear separation from HbA (Figure 1C).
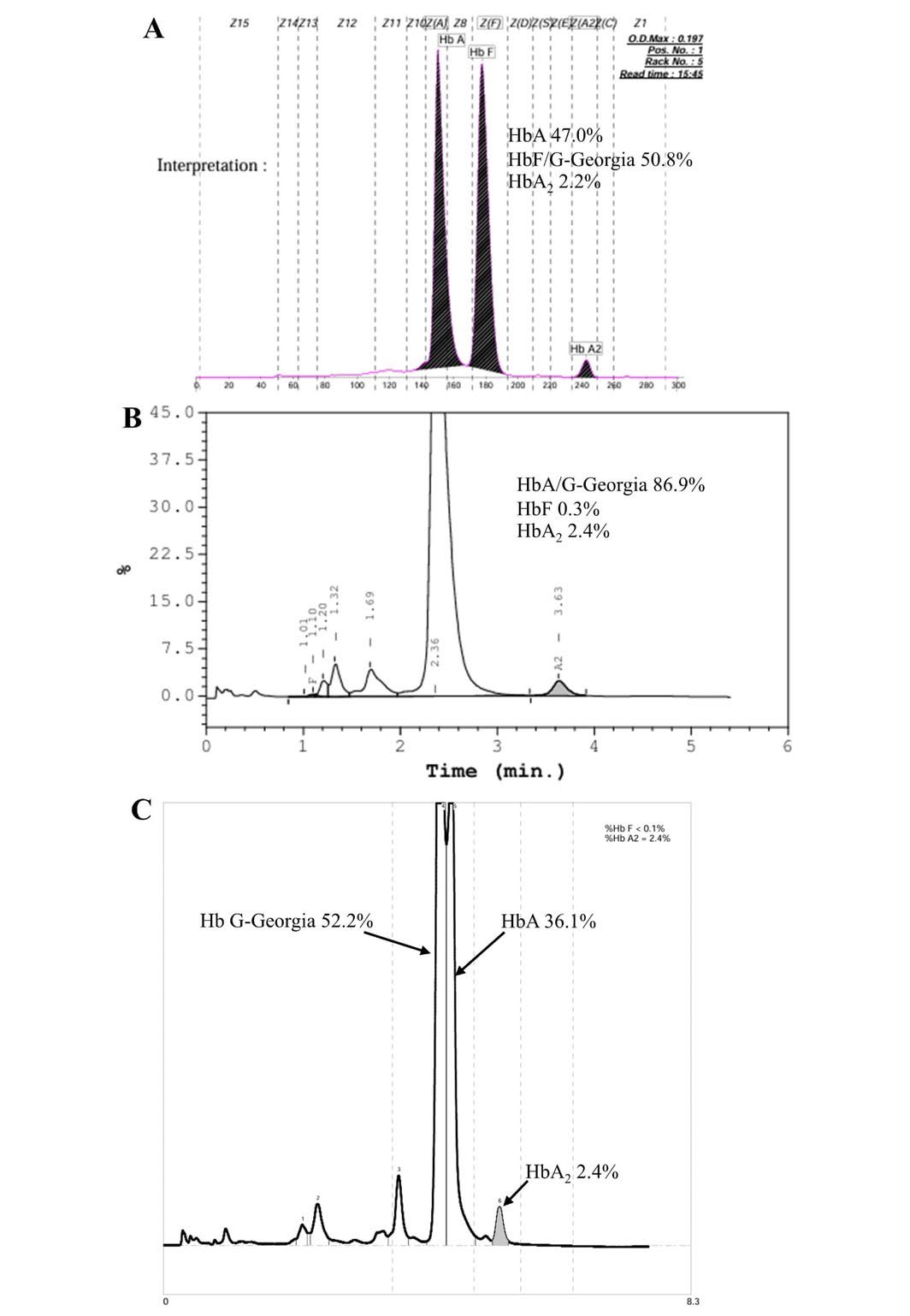
Figure 1. The CE electropherogram (A), HPLC chromatogram (B), and PR-HPLC chromatogram (C) of the case with compound heterozygosity for αGGα/--SEA.
To confirm the suspected mutation, a thalassemia gene panel targeting the coding regions of the HBA1, HBA2, and HBB genes was analyzed by NGS (BGI Group, Shenzhen, China) as previously described (Li et al., 2021). The CCG (Pro) to CUG (Leu) mutation at codon 95 of the α2-globin gene leading to the Hb G-Georgia (HBA2:c.287C>T) was observed as shown in Figure 2. The previous study found that the heterozygosity for Hb G-Georgia did not cause any clinical or hematologic effects, and the level of Hb G-Georgia was below 25%. However, this case had a microcytic hypochromic anemia (MCV 64.4 fL and MCH 20.0 pg). Therefore, her genomic DNA was extracted from the blood sample using the NucleoSpin® kit (Macherey-Nagel, KG., Duren, Germany) following the manufacturer’s instructions. Then, a single-tube multiplex real-time PCR with EvaGreen and high-resolution melting (HRM) analysis for the diagnosis of α0-thalassemia --SEA, --THAI, and --CR deletions was performed as described previously (Ruengdit et al., 2023). The analysis revealed a positive for α0-thalassemia --SEA type deletion. Thus, she was diagnosed as compound heterozygosity for Hb G-Georgia/ α0-thalassemia --SEA deletion (αGGα/--SEA).
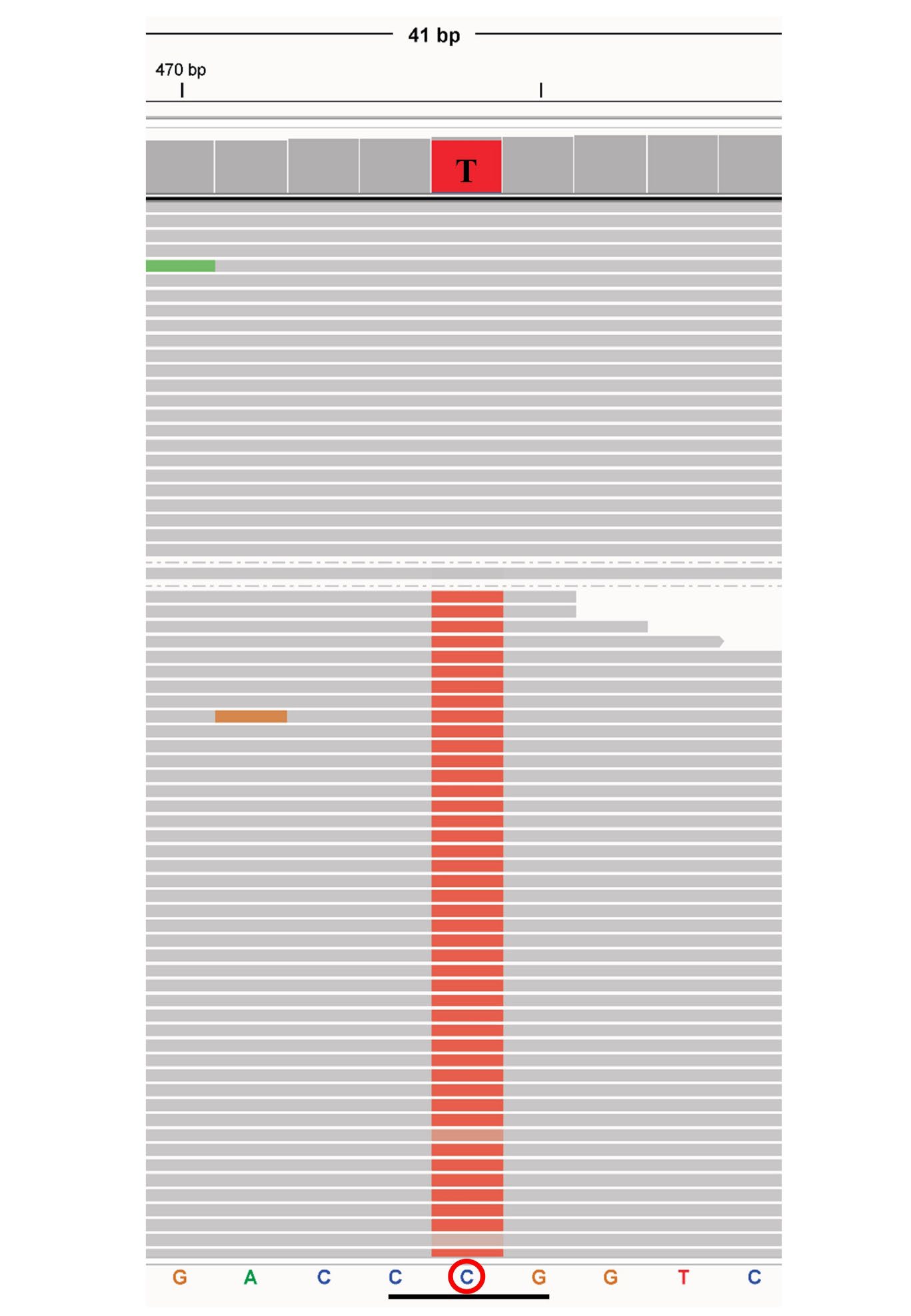
Figure 2. Representative of NGS results exported from the integrative genomics viewer (IGV) of Hb G-Georgia (HBA2:c.287C>T).
DISCUSSION
Hb G-Georgia was first observed in a 12-year-old Negro male orphan by Huisman et al. (1970). The second case was found in a Portuguese patient with lymph node tuberculosis, characterized by anemia, hemolytic crises, and moderate splenomegaly in 1975. The electrophoretic mobility of this abnormal Hb was slower than Hb S, and structural analysis revealed a leucine residue replacing a proline residue in position 95 in the α-globin chain (North et al., 1975). Another case was a Turkish woman in Amsterdam and the disorder was characterized by iron-resistant hypochromic microcytic anemia, splenomegaly, and increased hemoglobin degradation (van Houte et al., 1986). Moreover, the double heterozygosity for Hb G-Georgia/HbC (HBB:c.19G>A) and Hb G-Georgia/Hb Montreal II (β142 (-C)) were reported in a 12-month-old black female and a 25-year-old Caucasian man, respectively (Reynolds et al., 1992; Medri et al., 2022). In northeastern Thailand there had been a report of compound heterozygosity for Hb G-Georgia/α0-thalassemia --SEA type deletion in an adult male who had mild anemia (total Hb 114 g/L) with microcytic and hypochromic red cells (MCV 57.8 fL and MCH 18.2 pg, respectively) (Srivorakun et al., 2018) while in southern Thailand, there had been a report of double heterozygosity for Hb G-Georgia/HbE (HBB:c.79G>A) in an adult female (Tepakhan et al., 2024). Here we report the first case of compound heterozygosity for Hb G-Georgia/a0-thalassemia --SEA type deletion in northern Thailand. This co-inheritance led to her clinical symptoms of fatigue and dyspnea. Her hematological features revealed also a moderate anemia (total Hb 99 g/L) with microcytic and hypochromic red cells (MCV 64.4 fL and MCH 20.0 pg, respectively).
Heterozygosity for Hb G-Georgia is generally asymptomatic with Hb levels typically below 25%. However, when inherited with other mutations such as α0-thalassemia, more severe hematologic manifestations can occur. In this case, Hb G-Georgia level analyzed by CE and PR-HPLC was higher than 25% and that could be explained by the decrease of normal α-globin gene synthesis (Srivorakun et al., 2018; Tepakhan et al., 2024). Therefore, co-inheritance of Hb G-Georgia and α⁰-thalassemia --SEA type deletion led to microcytic hypochromic anemia despite normal iron status. Moreover, the diagnostic process revealed challenges in identifying Hb G-Georgia using HPLC or PR-HPLC due to its co-elution with HbA on HPLC chromatogram and did not clearly separate from HbA on PR-HPLC chromatogram. However, the CE proved more effective for separating and quantifying Hb G-Georgia in this case. The NGS confirmed the point mutation at codon 95 of the HBA2 gene, while α0-thalassemia --SEA deletion was detected by a single-tube multiplex real-time PCR with EvaGreen and HRM analysis. Therefore, this case highlights the diagnostic complexity of Hb G-Georgia and emphasizes the importance of integrating clinical and hematological manifestations with laboratory techniques for accurate diagnosis and improved patient management.
CONCLUSION
We report on a 62-year-old Thai woman with fatigue, dyspnea, pale without hepatosplenomegaly and blood transfusion history. Later, she was diagnosed as compound heterozygosity for Hb G-Georgia and α0-thalassemia --SEA deletion (αGGα/--SEA). This is the first case of Hb G-Georgia detected in northern Thailand. This study emphasizes the diagnostic challenges of Hb G-Georgia and the benefits of combining hematological and clinical features with laboratory techniques for precise diagnosis and improved patient care.
ACKNOWLEDGMENTS
The authors thank the technicians at Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand for their assistance.
ETHICAL CONSIDERATIONS
This study was approved by the Ethics Committee of the Faculty of Associated Medical Sciences, Chiang Mai University, Thailand (AMSEC-68EM-014).
AUTHOR CONTRIBUTIONS
Chedtapak Ruengdit and Manoo Punyamung assisted in conducting the experiments. Moe Theingi wrote the manuscript. Sakorn Pornprasert designed and conducted all the experiments and wrote the manuscript. All authors have read and approved of the final manuscript.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
Fucharoen, S. and Winichagoon, P. 1987. Hemoglobinopathies in southeast Asia. Hemoglobin. 11(1): 65-88.
He, J., Song, W., Yang, J., Lu, S., Yuan, Y., Guo, J., Zhang, J., Ye, K., Yang, F., Long, F., et al. 2017. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: The Dai nationality, China. Genetics in Medicine. 19(9): 1022-1031.
Huisman, T.H.J., Adams, H.R., Wilson, J.B., Efremov, G.D., Reynolds, C.A., and Wrightstone, R.N. 1970. Hemoglobin G Georgia or alpha 2-95 Leu (G-2) beta-2. Biochimica et Biophysica Acta (BBA) – Protein Structure. 200(3): 578-580.
Li, R., Shen, X., Chen, H., Peng, D., Wu, R., and Sun, H. 2021. Developmental validation of the MGIEasy signature identification library prep kit, an all-in-one multiplex system for forensic applications. International Journal of Legal Medicine. 135: 739-753.
Medri, C., Méndez, A., Hammerer-Lercher, A., Rovó, A., and Angelillo-Scherrer, A. 2022. Unstable hemoglobin Montreal II uncovered in an adult with unexplained hemolysis exacerbated by a presumed viral infection: A case report. Journal of Medical Case Reports. 16: 145.
North, M.L., Garel, M.C., Thillet, J., Oberling, F., Lang, J.M., Mayer, S., and Rosa, J. 1975. A new case of hemoglobin G Georgia (author's transl). Nouvelle Revue Francaise D'hematologie. 15(4): 460-467.
Reynolds, S., Miller, C., King, R., and Lazarchick, J. 1992. Hemoglobin C--G-Georgia double heterozygosity: A case report. Annals of Clinical Laboratory Sciences. 22(6): 414-416.
Ruengdit, C., Punyamung, M., Intasai, N., and Pornprasert, S. 2023. Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of alpha0-thalassemia--SEA,--THAI, and--CR type deletions. PLoS One. 18(11): e0293838.
Srivorakun, H., Singha, K., Fucharoen, G., Sanchaisuriya, K., and Fucharoen, S. 2014. A large cohort of hemoglobin variants in Thailand: Molecular epidemiological study and diagnostic consideration. PLoS One. 9(9): e108365.
Srivorakun, H., Singha, K., Fucharoen, G., and Fucharoen, S. 2018. Novel interactions of two α-Hb variants with SEA deletion α(0)-thalassemia: Hematological and molecular analyses. Hematology. 23(3): 187-191.
Tepakhan, W., Kanjanaopas, S., Sreworadechpisal, K., Penglong, T., Sripornsawan, P., Wangchauy, C., Nokkong, C., Kongkan, C., and Buathong, S. 2024. Molecular epidemiology and hematological profiles of hemoglobin variants in southern Thailand. Scientific Reports. 14: 9255.
van Houte, D.P., van den Ende, A., Statius van Eps, L.W., Giordano, P.C., and
Bernini, L.F. 1986. Hemoglobin G Georgia in a Turkish family in the Netherlands. Nederlands Tijdschrift Voor Geneeskunde. 130(8): 360-363.
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand. https://cmuj.cmu.ac.th
Moe Theingi1, Chedtapak Ruengdit2, Manoo Punyamung2, and Sakorn Pornpraset1, *
1 Division of Clinical Microscopy, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
2 Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
Corresponding author: Sakorn Pornpraset, E-mail: sakornmi001@gmail.com; sakorn.pornprasert@cmu.ac.th
ORCID ID: Sakorn Pornpraset: https://orcid.org/0000-0003-0280-5569
Total Article Views
Editor: Decha Tamdee,
Chiang Mai University, Thailand
Article history:
Received: May 2, 2025;
Revised: July 31, 2025;
Accepted: August 6, 2025;
Online First: August 20, 2025