 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Hb Westmead and Its Combinations with Other Forms of Thalassemia or Hemoglobinopathy in Northern Thailand
Thunyaluk Komrapit, Chedtapak Ruengdit, Manoo Punyamung, and Sakorn Pornprasert*Published Date : August 13, 2025
DOI : https://doi.org/10.12982/NLSC.2025.063
Journal Issues : Number 4, October-December 2025
Abstract Hemoglobin Westmead (Hb WM) (α122(H5) His>Gln) (HBA2: c.369C>G) is an α-globin variant that is misidentified by conventional Hb analysis techniques due to its inability to be distinguished from HbA. While Hb WM has been regarded as a silent Hb variant, its potential to produce clinically significant phenotypes when co-inherited with thalassemia or other hemoglobinopathies remains uncertain. The objective of this study was to identify Hb WM and its combinations with other forms of thalassemia or hemoglobinopathies in Northern Thailand. Hb WM was identified by using targeted next-generation sequencing (NGS). Routine hemoglobin analysis was also conducted using high-performance liquid chromatography (HPLC) and/or capillary electrophoresis (CE) techniques. The α0-thalassemia --SEA, --THAI, and --Chiang Rai type deletions were identified using real-time polymerase chain reaction combined with high-resolution melting (HRM) analysis. The Hb WM was found in 74 of 2,773 samples (2.6%), one sample was identified as homozygosity for Hb WM, while 73 samples were classified as heterozygosity for Hb WM. Nine distinct genotypes were analyzed, revealing variability in clinical and hematological characteristics based on their combinations. The knowledge of Hb WM and its combinations is useful in thalassemia screening, genetic counseling, control, prevention, and treatment.
Keywords: Hb Westmead, Hematological parameters, Hemoglobinopathy, Next-Generation sequencing, Thalassemia
Citation: Komrapit, T., Ruengdit, C., Punyamung, M., and Pornprasert, S. 2025. Hb Westmead and its combinations with other forms of thalassemia or hemoglobinopathy in Northern Thailand. Natural and Life Sciences Communications. 24(4): e2025063.
INTRODUCTION
Alpha-thalassemia is a genetic disorder highly prevalent in Southeast Asian countries, particularly Thailand (Chaibunruang et al., 2013). This disorder results from different degrees of α-globin chain deficiency, contingent upon the quantity of defective α-globin genes, which can arise from deletions or point mutations. A total deficit in α-globin synthesis (α0-thalassemia) occurs when both adjacent α-globin genes on chromosome 16p13.3 are deleted. The deletion or mutation of a single α-globin gene (α⁺-thalassemia) results in a partial decrease in α-globin synthesis (Muncie and Campbell, 2009). The interaction between α0-thalassemia and α⁺-thalassemia leads to a moderate form of α-thalassemia named HbH disease, which is characterized by moderate anemia along with substantial microcytosis. The most severe form occurs when all four α-globin genes are nonfunctional, resulting in hydrops fetalis, a condition that usually leads to fetal demise (Tamary and Dgany, 2005). Whereas deletions are the most frequent cause of α-thalassemia, certain point mutations and α-globin variants can also lead to thalassemia-like phenotype and minimal or no noticeable hematologic abnormalities. Generally, a compound heterozygosity for α0-thalassemia with non-deletional mutations tends to cause a more significant reduction in α-globin chain production than those with deletions. Consequently, individuals with non-deletional HbH disease typically exhibit more severe anemia than those with the deletional form (Piel and Weatherall, 2014).
Hb Westmead (WM) is caused by point mutation in α2-globin gene (HBA2), specifically a C→G substitution at codon 122 (HBA2: c.369C>G). This mutation results in the amino acid change from histidine to glutamine at position 122 of the α-globin chain (α122(H5) His→Gln)). It cannot be identified by routine Hb analysis methods, including a high-performance liquid chromatography (HPLC) and a capillary electrophoresis (CE) system (Jiang et al., 2020a). Hb WM was initially characterized by Fleming et al. citation from 1980 regarding a family that migrated from Guangdong, Southern China, to Australia (Fleming et al., 1980). This variant is commonly found in Chinese population (Xiong et al., 2010; Jiang et al., 2020a). Although Hb WM has been considered as a silent Hb variant or as an α+-thalassemia allele (Jiang et al., 2020a), its interaction with α0- or β0-thalassemia may exacerbate anemia due to imbalanced globin chain synthesis. This suggests a need for advanced molecular diagnostics in routine screening programs to better understand and manage potential interactions. In Thailand, the first case of Hb WM was reported in a 2-year-old Thai boy from Uttaradit Province who had presented with severe anemia at the age of 4 months and had diagnosed as double heterozygosity for Hb WM and β0-thalassemia (αα/αWMα, β0/βA) (Viprakasit et al., 2014). Therefore, aims of this study were to investigate the frequency of Hb WM in Northern Thailand, as Hb WM is commonly found in China but has been rarely reported in Thailand. The genotypes and phenotypes of Hb WM and its combination forms with other thalassemia or hemoglobinopathies were also described.
MATERIAL AND METHODS
Blood samples and thalassemia diagnosis
This study was approved by the Ethics Committee of the Faculty of Associated Medical Sciences, Chiang Mai University, Thailand (AMSEC-67EM-036). Blood samples anti-coagulated with ethylenediamine tetra acetic acid (EDTA) and the data of red cell indices, including the red blood cell counts (RBCs), total Hb concentration, packed cell volume (PCV), mean corpuscular volume (MCV), mean corpuscular Hb (MCH), and mean corpuscular Hb concentration (MCHC) measured by automated cell counters were submitted from public and private hospitals in Northern Thailand to the Associated Medical Sciences-Clinical Service Center (AMS-CSC), Chiang Mai University, Chiang Mai, Thailand for routine thalassemia diagnosis for standard thalassemia diagnosis due to positive outcomes in thalassemia screening (MCV <80 fL, MCH <27 pg, and/or positive Dichlorophenol Indophenol Precipitation (DCIP) test). At the AMS-CSC, a high-performance liquid chromatography (HPLC) (VARIANT β-thalassemia Short Program; Bio-Rad Laboratories, Hercules, CA, USA) and/or a capillary electrophoresis (CE) system (Capillarys 2 Flex piercing, Sebia, Evry, France) were employed to quantify HbA2 for β-thalassemia detection and to identify hemoglobinopathies. Genomic DNA was extracted from the whole blood sample using the Chelex method (Chelex 100 Resin, Sigma, CA, USA) (Walsh et al., 1991). A single-tube multiplex real-time PCR with EvaGreen and high-resolution melting (HRM) analysis for the diagnosis of α0-thalassemia --SEA, --THAI, and --Chiang Rai deletions was performed as described previously (Ruengdit et al., 2023). In addition, to assay the other types of thalassemia mutations and hemoglobinopathies, the NGS panel targeting the coding regions of the HBA1, HBA2, and HBB genes was performed using a Thalassemia Gene Detection Kit (BGI Group, Shenzhen, China) as described in the previous study (He et al., 2017). Sequence reads were aligned to the hg19 build of the human genome, and sequence variants were interpreted using THACARE HALOS analysis software (BGI Group).
RESULTS
From August to November 2023, a total of 2,773 EDTA anticoagulated blood samples were subjected to detect α- and β-globin gene variants using NGS. The sequencing data of Hb Westmead identified by NGS is shown in Figure 1. Based on the NGS system and routine thalassemia diagnosis methods, Hb WM was found in 74 (2.6%) individuals, including 9 different genotypes. The characteristics and hematological parameters of 9 different genotypes are shown in Table 1. The subjects with heterozygosity for Hb WM (αα/αWMα & βA/βA), compound heterozygosity for Hb WM and Hb CS or α+-thalassemia 3.7 kb deletion (αCSα/αWMα & βA/βA, or -α3.7/αWM α & βA/βA), and double heterozygosity for Hb WM and HbE (αα/αWMα & βE/βA) had mean levels of RBCs, total Hb, MCV, MCH, and MCHC closed to or within the reference ranges (Table 1). The normal levels of HbA, HbA2, and HbF were found also in subjects with heterozygosity for Hb WM (Figure 2A) and compound heterozygosity for Hb WM and α+-thalassemia 3.7 kb deletion (Figure 2D). In addition, peaks of Hb CS in C zone (0.7%) and HbE were observed on CE electropherograms of subjects with compound heterozygosity for Hb WM and Hb CS (Figure 2B) and double heterozygosity for Hb WM and HbE (Figure 2G), respectively. The normal levels of MCV and MCH (Table 1) and HbA, HbA2, and HbF (Figure 2I) were revealed in the subject with homozygosity for Hb WM, even thought she had the lowest levels of RBCs, total Hb, and PCV when compared with other thalassemia genotypes.
The subjects with compound heterozygosity for Hb WM and α0-thalassemia --SEA type deletion and double heterozygosity for Hb WM and β0-thalassemia had levels of total Hb, PCV, MCV, and MCH lower than the reference ranges (Table 1). Peaks of Hb Bart’s and Hb H were not observed on the CE electropherogram of subjects with compound heterozygosity for Hb WM and α0-thalassemia --SEA type deletion (Figure 2C) while the peak of elevated HbA2 (>4.0%) was found on the CE electropherogram of subjects with double heterozygosity for Hb WM and β0-thalassemia (Figure 2F). The lower levels of total Hb, PCV, MCV, and MCH (Table 1) and the of elevated HbA2 (>4.0%) were revealed also in subjects with compound heterozygosity for Hb WM and α+-thalassemia 3.7 kb deletion or α0-thalassemia --SEA type deletion who had a co-inheritance of β0-thalassemia (Figure 2E and 2H, respectively).
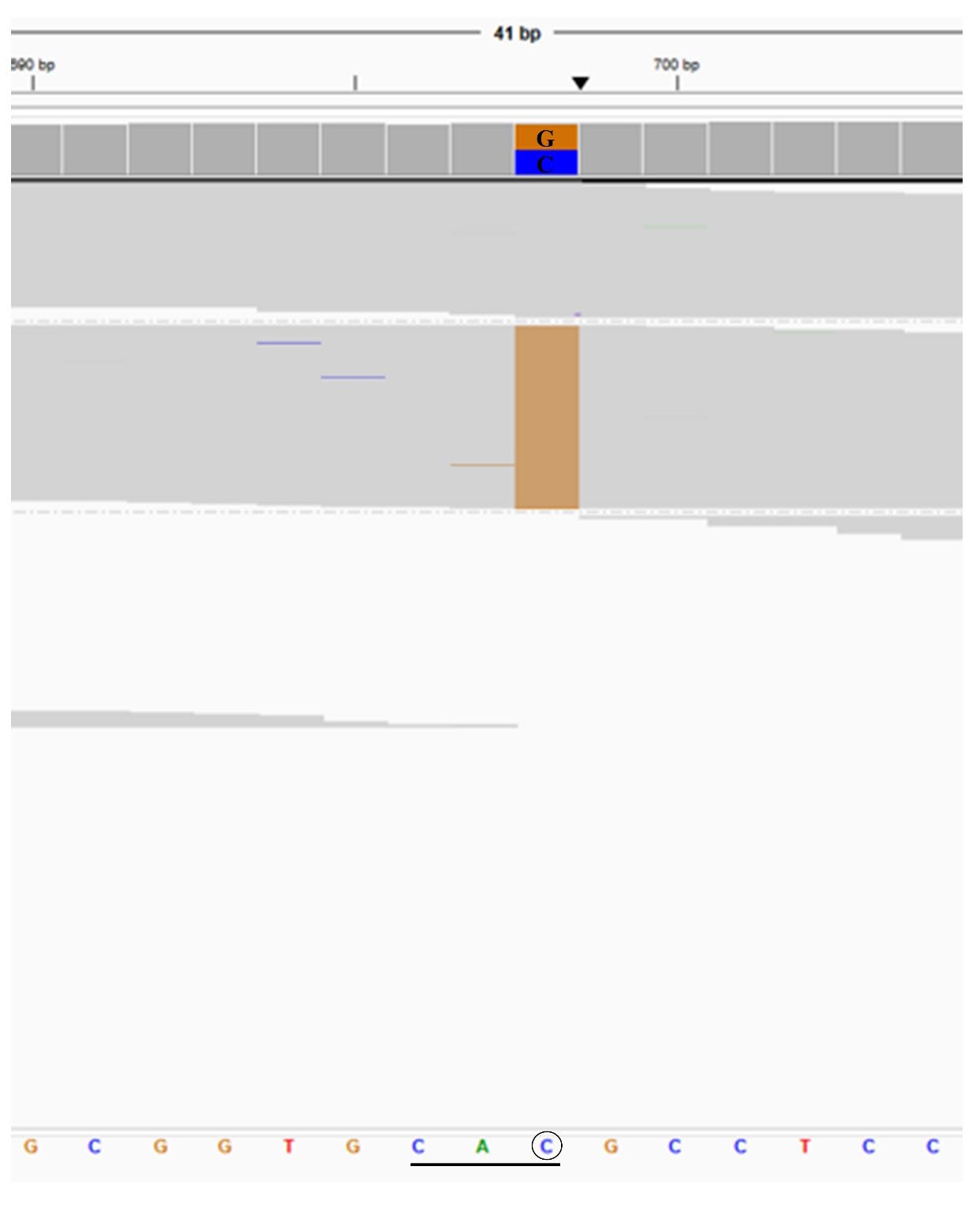
Figure 1. Representative of NGS results exported from the integrative genomics viewer (IGV) of the heterozygosity for Hb Westmead (HBA2: c.369C>G).
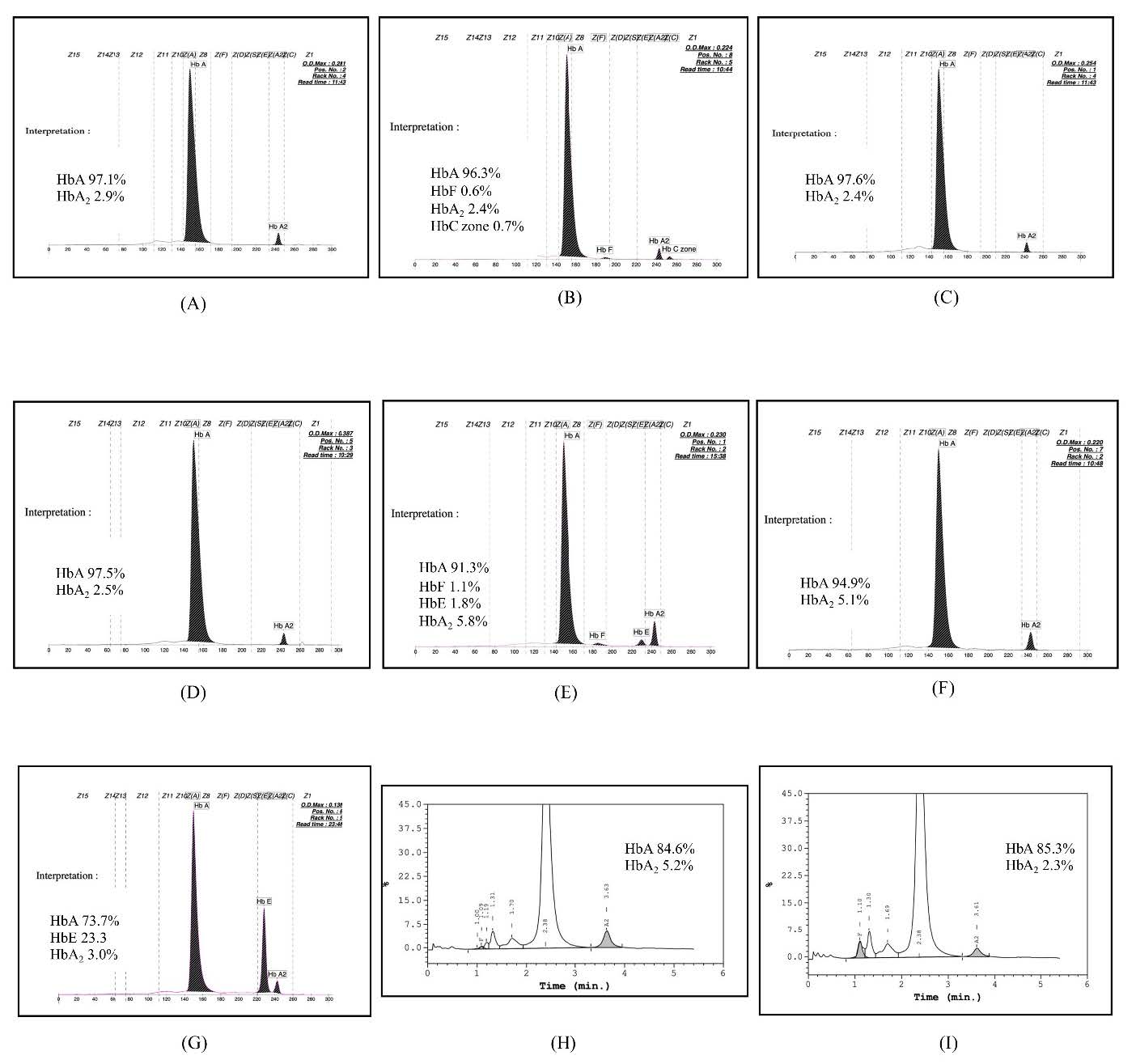
Figure 2. Representative of the HPLC chromatograms and/or CE electropherograms of nine genotypes, including αα/αWMα, bA/bA (A), αCSα/αWMα, bA/bA (B), --SEA/αWMα, bA/bA (C), -α 3.7/αWMα, bA/bA (D), -α 3.7/αWMα, b0/bA (E), αα/αWMα, b0/bA (F), αα/αWMα, bE/bA (G), --SEA/αWMα, b0/bA (H), and αWMα/αWMα, bA/bA (I).
Table 1. Characteristics, hemoglobin analysis and hematological parameters of Hb Westmead and its combinations.
|
Genotypes (N; Male/Female) |
Age (Years) (Ranges) |
Hematological parameters |
|
Hb analysis by HPLC or CE (%) |
|||||||
|
RBCs (x1012/L) |
Hb (g/dL) |
PCV (L/L) |
MCV (fL) |
MCH (pg) |
MCHC (g/L) |
|
HbA* |
HbA2 |
HbF |
||
|
αα/αWMα, bA/bA (32; 6/26) |
43.0 ± 21.5 (1 - 81) |
4.4 ± 1.1 (2.7 - 5.8) |
11.5 ± 3.0 (6.8 - 16.1) |
0.35 ± 0.08 (0.24 - 0.49) |
80.9 ± 9.7 (52 - 101) |
26.3 ± 3.9 (19.6 - 28.8) |
325 ± 18.5 (308 - 367) |
|
89.3 ± 5.8 (79.1 -98.3) |
2.9 ± 0.4 (1.7 - 3.4) |
0.4 ± 0.5 (0.0 - 2.0) |
|
--SEA/αWMα, bA/bA (5; 3/2) |
26.5 ± 22.1 (3 - 56) |
5.3 ± 1.4 (3.7 - 7.0) |
10.7 ± 3.7 (6.4 - 14.9) |
0.34 ± 0.09 (0.23 - 0.45) |
62.8 ± 3.0 (59 - 66) |
19.7 ± 2.4 (17.3 - 22.2) |
313 ± 26.5 (278 - 336) |
|
95.25 ± 4.5 (88.6 - 98.1) |
2.5 ± 0.6 (1.9 - 3.3) |
0.05±0.1 (0.0 - 0.2) |
|
αCSα/αWMα, bA/bA (2; 1/1) |
15 / 21 |
4.1 / 5.4 |
11.2 / 14.2 |
0.33 / 0.42 |
80 / 79 |
27.2 / 26.2 |
339 / 332 |
|
96.3 / 88.4 |
2.4 / 3.2 |
0.6 / 0.7 |
|
-α3.7/αWMα, bA/bA (8; 3/5) |
34.0 ± 18.5 (1 - 63) |
5.4 ± 0.6 (4.9 - 6.2) |
12.8 ± 2.6 (10.5 - 17.4) |
0.39 ± 0.06 (0.32 - 0.50) |
72.2 ± 8.0 (64 - 82) |
23.4 ± 3.2 (19.6 - 28) |
327 ± 15.1 (304 - 344) |
|
90.2 ± 6.1 (85.4 - 97.6) |
2.9 ± 0.5 (2.4 - 3.7) |
0.5 ± 0.5 (0.5 - 1.3) |
|
-α3.7/αWMα, b0/bA (1; 1/0) |
49.0 |
4.6 |
9.3 |
0.29 |
62 |
20.2 |
325 |
|
91.3 |
5.8 |
1.1 |
|
αα/αWMα, b0/bA (14; 3/11) |
39.0 ± 25.0 (3- 88) |
5.1 ± 1.0 (2.6 - 6.3) |
10.8 ± 0.8 (8.5 - 13.7) |
0.35 ± 0.02 (0.31 - 0.40) |
69.9 ±12.0 (58 - 99) |
22.0 ± 4.8 (17.7 - 32.9) |
313 ± 18.2 (294 - 348) |
|
86.6 ± 9.0 (69.7 - 97.4) |
5.1± 1.4 (3.7 - 7.3) |
3.2 ± 4.8 (0.0 - 14.1) |
|
αα/αWMα, bE/bA (10; 3/7) |
24.0 ± 11.1 (1 - 39) |
4.8 ± 0.7 (3.8 - 5.8) |
11.5 ± 2.1 (8.3 - 13.8) |
0.35 ± 0.06 (0.28 - 0.41) |
73.5 ± 4.1 (72 - 81) |
23.7 ± 1.6 (21.8 - 25.9) |
323 ± 16.4 (299 - 345) |
|
67.9 ± 6.2 (58.6 - 75.8) |
27.0 ± 1.9# (24.2 - 29.8) |
0.8 ± 1.2 (0.0 - 3.7) |
|
--SEA/αWMα, b0/bA (1;0/1) |
63 |
4.8 |
9.9 |
0.30 |
69 |
20.7 |
300 |
|
84.6 |
5.2 |
0.5 |
|
αWMα /αWMα, bA/bA (1;0/1) |
67 |
2.6 |
7.7 |
0.23 |
98 |
30.2 |
308 |
|
85.3 |
2.3 |
2.9 |
Note: Normal range: red blood cell counts (RBCs), 4.2 - 6.1x1012/L; total Hb, 12.0 -18.0 g/dL; packed cell volume (PCV), 0.37 - 0.52 L/L; mean corpuscular volume (MCV), 80 - 100 fL; mean corpuscular Hb (MCH), 27 - 31 pg; mean corpuscular Hb concentration (MCHC), 320 - 360 g/L; HbA >85%; HbA2 1.5 - 3.5%; HbF 0.0 - 1.0%
*HbA = HbA + Hb Westmead
#HbA2 = HbA2 + HbE
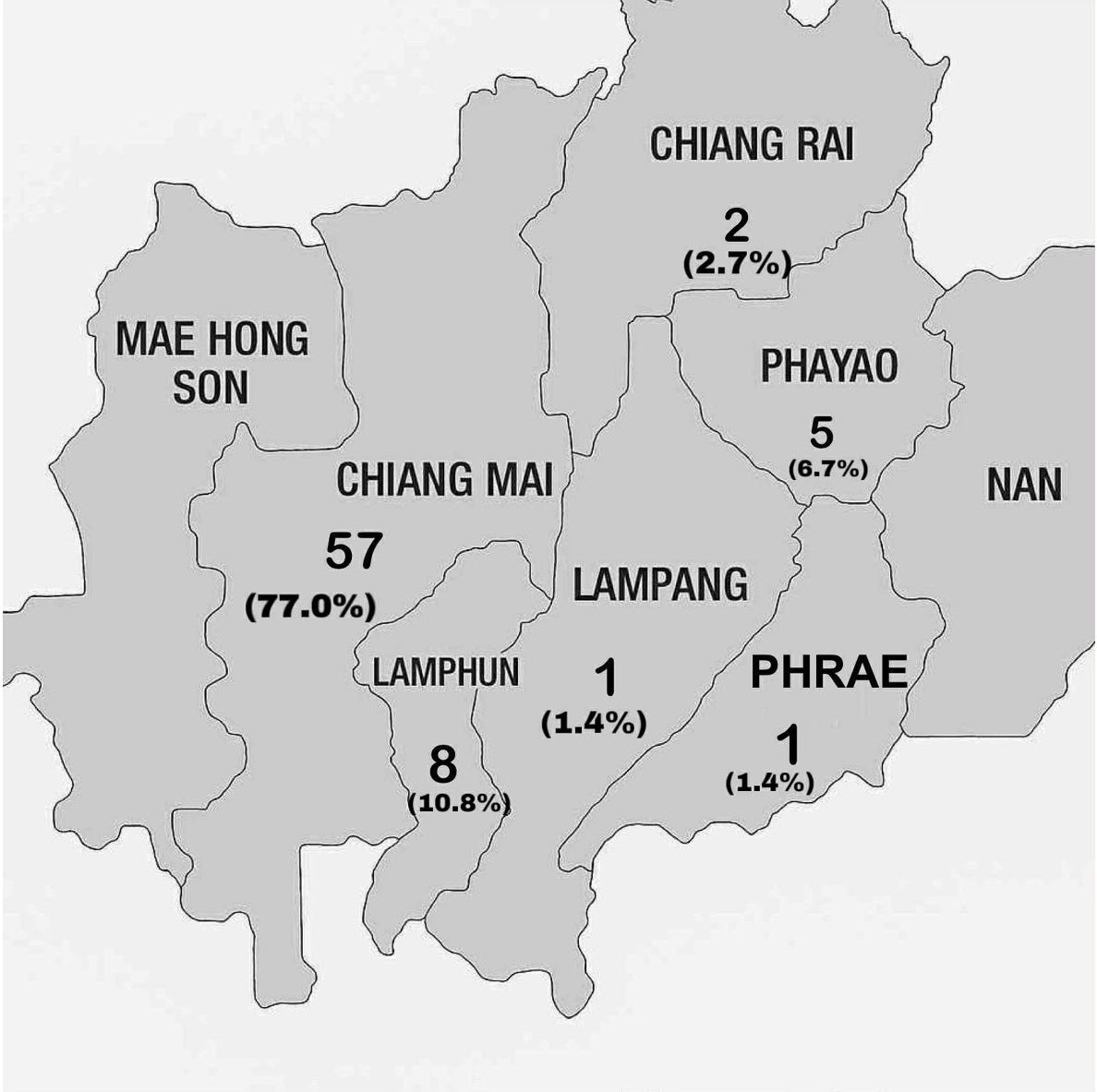
Figure 3. Geographical distribution of Hb WM samples.
DISCUSSION
This study reported for the first time of Hb WM and its combinations with other forms of thalassemia or hemoglobinopathy found in Northern Thailand. Among 2,773 subjects, Hb WM was found in 74 (2.6%) individuals (1 homozygosity and 73 heterozygosity). Figure 3 exhibits the geographical distribution of Hb WM samples. The majority of cases are found in Chiang Mai (77.0%), followed by Lamphun (10.8%), and Phayao (6.7%) provinces. The number of cases correlated with the amount of blood samples collected from each province. Thus, the allele frequency of Hb WM in this region was 1.35%, which is comparable to that in Southern China, where the frequency was 1.55%. It ranks as the fourth most prevalent α-thalassemia allele, following the --SEA deletion, -α3.7 deletion, and -α4.2 deletion in Southern China population (Xiong et al., 2010). Moreover, based on the NGS and routine laboratory diagnosis, the combination forms of Hb WM with other thalassemia and hemoglobinopathies could be classified into 9 different genotypes which presented different hematological and clinical features. The heterozygosity for Hb WM (αα/αWMα, βA/βA) had no clinical symptoms and their hematological parameters were found closely to or within the reference ranges. Thus, Hb WM has been regarded as a silent Hb variant or as an α+-thalassemia allele (Jiang et al., 2020a). Although, the 67-year-old woman who was homozygosity for Hb WM had severe anemia with total Hb 7.7 g/dL, her MCV (98 fL) and MCH (30.2 pg) were observed within the reference ranges which indicated that anemia in this patient may be due to acute blood loss or ineffective erythropoiesis (Crowther, 2021). However, the etiology of her anemia requires additional investigation. In addition, the compound heterozygosity for Hb WM and α+-thalassemia exhibited a lesser impact on hematological parameters compared to the homozygosity for α+-thalassemia and the heterozygosity for α0-thalassemia (Chaibunruang et al., 2013). Therefore, Hb WM is considered as a silent Hb variant rather than α+-thalassemia allele. Moreover, the peaks of Hb Bart’s and HbH were not observed on the CE electropherogram of subjects with compound heterozygosity for Hb WM and α0-thalassemia (Figure 2C). Even though, Hb WM mutation occurs in the same exon 3 of HBA2 gene as Hb Constant Spring (CS; CD142 T>C; HBA2: c.427 T>C), Hb Paksé (PS; CD142 A>T; HBA2: c.429 A>T), and Hb Quong Sze (Hb QS; CD125 T>C; HBA2: c.377 T>C), it does not affect the α-globin chain production and stabilization, and Hb formation. Thus, unlike Hb CS, Hb PS, and Hb QS, the compound heterozygosity for Hb WM and α0-thalassemia does not generate non-deletional HbH disease (Fucharoen et al., 1988; Viprakasit et al., 2002; Pornprasert et al., 2012a; Jiang et al., 2020b; Zhong et al., 2023). However, we emphasize that if one partner of a couple has tested positive for α0-thalassemia, the other should be subjected to detailed screening for these non-deletional alleles using molecular analysis.
The previous study demonstrated that the co-inheritance of α-thalassemia ameliorated the clinical phenotype of severe β-thalassemia (Olivieri, 1999). However, in the present study, the subjects with double heterozygosity for Hb WM and β0-thalassemia (αα/αWMα, β0/βA) or co-inheritance of compound heterozygosity for Hb WM and α+-thalassemia 3.7 kb deletion and β0-thalassemia trait (-α3.7/αWMα, β0/βA) had mild to moderate anemia with microcytic and hypochromic red blood cells as usually found in subjects with heterozygosity for β-thalassemia (Pornprasert et al., 2012b). Consistency with the previous study showed that Hb WM did not improve the severe phenotype of β-thalassemia major (β0/β0) (Galanello and Origa., 2010). Conversely, Wong et al. (2014) elucidated that the presence of Hb WM, alongside the α0-thalassemia deletion, mitigates the excess of α-globin chains, resulting in a less severe condition in a patient with --SEA/αWMα & β41/42/β- 28 (Wong et al., 2014). Furthermore, the co-inheritance of α-thalassemia with Hb E results in a marked reduction of HbA2/E level (<25%) (Pornprasert et al., 2012b). However, the current study showed that Hb WM did not reduce the HbA2/E levels in the subject with double heterozygosity for Hb WM and Hb E (αα/αWMα, βE/βA) (Figure 2G). A minor peak of HbE (1.8%) on the CE electropherogram of an individual with compound heterozygosity for Hb WM and α+-thalassemia 3.7 kb deletion, who also had co-inheritance of β0-thalassemia (Figure 2E), may have resulted from having blood transfusions from a donor with HbE prior to Hb analysis (Pornprasert and Jaiping, 2014). Nevertheless, the HbE transfusion residue has not been molecularly assessed and validated in the present investigation.
The current study has limitations. Firstly, our study aimed to analyze the genotype and phenotype of Hb WM and its combinations with other forms of thalassemia or hemoglobinopathies in Northern Thailand; therefore, samples were primarily focused on individuals with positive outcomes in thalassemia screening (MCV <80 fL, MCH <27 pg, and/or DCIP test) and hospital-based sampling. Consequently, the manuscript underestimates the frequency of Hb WM, as it excludes people with normal red cell indices and hemoglobin levels. Thus, the prevalence of Hb WM in the Northern Thai population necessitates screening of the healthy general population. Secondly, 74 individuals with Hb WM, nine in combination with other globin gene mutations, may be insufficient in number to establish any valid relationships between genotype and phenotype. Furthermore, the number of Hb WM cases identified in the present study correlated with the amount of blood samples obtained from each province. Therefore, further large-scale population studies along the Northern Thailand are required to validate these findings.
CONCLUSION
In conclusion, we identified Hb WM mutation and its combinations with other forms of thalassemia or hemoglobinopathy in Northern Thailand. The allele frequency of Hb WM in Northern Thailand was 1.35%. The frequency of Hb WM is significantly underestimated in the population, as the most employed separation technologies fail to distinguish it from HbA. In addition, Hb WM could be co-inherited with other thalassemia or hemoglobinopathies, resulting in a total of 9 different genotypes. Although Hb WM is a silent mutation, the clinical and hematological features varied with their combination forms. Identifying Hb WM is crucial for accurate diagnosis, effective clinical treatment, and appropriate genetic counseling, especially in regions with a high prevalence of thalassemia and hemoglobinopathies, such as Northern Thailand. Thus, the molecular diagnosis of Hb WM can prevent misdiagnosis and enhance methodologies for carrier screening and prenatal diagnosis.
ACKNOWLEDGMENTS
The authors thank technicians in the Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand for their help and assistance.
AUTHOR CONTRIBUTIONS
TK, CR, MP, and SP were involved in the conception of the study. TK and CR performed the data collection and the analyses. TK, CR, and SP were involved in the interpretation of the results. TK and SP drafted the manuscript. CR and MP gave critical feedback on the manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
Chaibunruang, A., Prommetta, S., Yamsri, S., Fucharoen, G., Sae-Ung, N., Sanchaisuriya, K., and Fucharoen, S. 2013. Molecular and hematological studies in a large cohort of α(0)-thalassemia in northeast Thailand: Data from a single referral center. Blood Cells, Molecules and Diseases. 51(2): 89-93.
Crowther, M. and Podolak-Dawidziak M. 2021. Anemia: General considerations. McMaster Textbook of Internal Medicine. Kraków: Medycyna Praktyczna. https://empendium.com/mcmtextbook-sae/chapter/B78.II.15.1.?rfmcm Accessed May 07, 2025.
Fleming, P.J., Hughes, W.G., Farmilo, R.K., Wyatt, K., and Cooper, W.N. 1980. Hemoglobin Westmead alpha 2 122(H5)His replaced by Gln beta 2: A new hemoglobin variant with the substitution in the alpha 1 beta 1 contact area. Hemoglobin. 4(1): 39-52.
Fucharoen, S., Winichagoon, P., Pootrakul, P., Piankijagum, A., and Wasi, P. 1988. Differences between two types of Hb H disease, α-thalassemia 1/α-thalassemia 2 and α-thalassemia 1/Hb constant spring. Birth Defects Original Article Series. 23(5B): 309-315.
Galanello, R. and Origa, R. 2010. Beta-thalassemia. Orphanet Journal of Rare Diseases. 5: 11.
He, J., Song, W., Yang, J., Lu, S., Yuan, Y., Guo, J., Zhang, J., Ye, K., Yang, F., Long, F., et al. 2017. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: The Dai nationality, China. Genetics in Medicine. 19(9): 1022-1031.
Jiang, F., Ju, A. P., Li, J., Chen, G.L., Zhou, J.Y., Tang, X.W., Zuo, L.D., and Li, D.Z. 2020a. Hb Westmead (HBA2: c.369C>G): Hematological characteristics in heterozygotes with and without α0-thalassemia. Hemoglobin. 44(2): 153-155.
Jiang, F., Xu, L.L., Chen, G.L., Zhou, J.Y., Li, J., Tang, X.W., Zuo, L.D., and Li, D.Z. 2020b. Hematological characteristics of Hb Constant Spring (HBA2: c.427T>C) carriers in mainland China. Hemoglobin. 44(2): 86-88.
Muncie, H.L. Jr. and Campbell, J. 2009. Alpha and beta thalassemia. American Family Physician. 80(4): 339-344.
Olivieri, N.F., 1999. The β-thalassemias. New England Journal of Medicine. 341(2): 99-109.
Piel, F.B. and Weatherall, D.J. 2014. The α-thalassemias. New England Journal of Medicine. 371(20): 19081916.
Pornprasert, S. and Jaiping, K. 2014. Blood transfusion from a Hb E trait donor can affect β-thalassemia diagnosis. Hemoglobin. 38(4): 295-298.
Pornprasert, S., Panyasai, S., and Treesuwan, K. 2012a. Unmasking Hb Paksé (codon 142, TAA>TAT, α2) and its combinations in patients also carrying Hb Constant Spring (codon 142, TAA>CAA, α2) in northern Thailand. Hemoglobin. 36(5): 491-496.
Pornprasert, S., Treesuwan, K., Punyamung, M., and Kongthai, K. 2012b. Hb A2/E levels found in co-inheritance with the α-thalassemia-1 --SEA/type deletion and either Hb E or β-thalassemia. Hemoglobin. 36(4): 381-387.
Ruengdit, C., Punyamung, M., Intasai, N., and Pornprasert, S. 2023. Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of alpha0-thalassemia--SEA, --THAI, and --CR type deletions. PLoS One. 18(11): e0293838.
Tamary, H. and Dgany, O. 1993. Alpha-thalassemia. In: Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., and Amemiya, A, editors. Current Molecular Medicine. Seattle: University of Washington. [accessed 2025 May 7]. https://www.ncbi.nlm.nih.gov/books/NBK1435/
Viprakasit, V., Ekwattanakit, S., Chalaow, N., Riolueang, S., Wijit, S., Tanyut, P., Chat-Uthai, N., and Tachavanich, K. 2014. Clinical presentation and molecular identification of four uncommon alpha globin variants in Thailand: Initiation codon mutation of α2-globin gene (HBA2:c.1delA), donor splice site mutation of α1-globin gene (IVS1-1, HBA1:c.95+1G>A), Hemoglobin Queens Park/Chao Pra Ya (HBA1:c.98T>A), and Hemoglobin Westmead (HBA2:c.369C>G). Acta Haematologica. 132(1): 88-94.
Viprakasit, V., Tanphaichitr, V.S., Pung-Amritt, P., Petrarat, S., Suwantol, L., Fisher, C., and Higgs, D. R. 2002. Clinical phenotypes and molecular characterization of Hb H-Paksé disease. Haematologica. 87(2): 117-125.
Walsh, P.S., Metzger, D.A., and Higuchi, R. 1991. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 10(4): 506-513.
Wong, W.S., Chan, A.Y., Yip, S F., and Ma, E.S. 2014. Thalassemia intermedia due to co-inheritance of β0/β+-thalassemia and (--SEA) α-thalassemia/Hb Westmead [α122(H5)His>Gln (α2)] in a Chinese family. Hemoglobin. 38(3): 215-220.
Xiong, F., Sun, M., Zhang, X., Cai, R., Zhou, Y., Lou, J., Zeng, L., Sun, Q., Xiao, Q., Shang, X., et al. 2010. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clinical Genetics. 78(2): 139-148.
Zhong, Z., Guan, Z., Chen, D., Zhong, G., He, H., Yang, K., and Chen, J. 2023. Molecular analysis and clinical significance of hemoglobin Quong Sze in Huizhou City, southern China. Taiwanese Journal of Obstetrics and Gynecology. 62(5): 709-712.
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand. https://cmuj.cmu.ac.th
Thunyaluk Komrapit1, Chedtapak Ruengdit2, Manoo Punyamung2, and Sakorn Pornprasert1, *
1 Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
2 Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
Corresponding author: Sakorn Pornprasert, E-mail: sakornmi001@gmail.com
ORCID: Sakorn Pornprasert: https://orcid.org/0000-0003-0280-5569
Total Article Views
Editor: Decha Tamdee,
Chiang Mai University, Thailand
Article history:
Received: May 7, 2025;
Revised: July 18, 2025;
Accepted: July 22, 2025;
Online First: August 13, 2025