 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Production of Encapsidated RNA Particles as a Working Standard in Detecting Foodborne Viruses in Oysters
Uraiwan Intamaso*, Palatip Chutoam, Suthasinee Jinda, and Supannee LethochavalitPublished Date : April 3, 2023
DOI : https://doi.org/10.12982/NLSC.2023.034
Journal Issues : Number 2, April-June 2023
Abtract Monitoring foodborne viruses via nucleic acid amplification tests rely on stable RNA standards to obtain reliable testing. This study aimed to produce RNA-based standard reagents for hepatitis virus (HAV) or norovirus detections which relies on viral-like particle (VLP) technology. Using a plasmid packaging system, plasmids containing DNA encoding Qβcoat protein (CP) monomer and the VP1 gene of viruses were co-transformed into E. coli host cells. In cell lysates, expressed CP was characterized by western blot and the whole icosahedral formation of VLPs was proved by electron microscope analysis. Encapsidated RNAs were measured and assessed as a standard by a two-step reverse transcription recombinase polymerase amplification (RT-RPA). Our results showed that CP has a distinguished protein band with a molecular weight of 14.5 kDa but a few variabilities of particle size were visualized. When adjusting the pH of the lysate to lower than 6, a more intense protein band and substantial particles with homogenous particle size were observed. These VLPs were found to enclose HAV and norovirus RNA contents to 1.2×107 copies/ng and 1.9×107 copies/ng, respectively. When analyzed by RT-RPA, linear regression analysis confirmed the alternative application of RNAs enclosed in VLPs to naked RNA synthesized from in vitro transcription. Using the E. coli expression system to produce Qβ VLPs allows cost-effective production and, therefore, can be implemented in laboratories with basic equipment. These encapsidated RNAs may become an ideal “standard” for detecting foodborne viruses via a molecular test in food and clinical samples.
Keywords: Molecular testing, Nanoparticles, Nucleic amplification, RNA standards, Viral-like particles
Funding: This research project was financially supported by Agricultural Research Development Agency (Public Organization) or "ARDA," grant number 6305031040.
Citation: Intamaso, U., Chutoam, P., Jinda, S., and Lethochavalit, S. 2023. Production of encapsidated RNA particles as a working standard in detecting foodborne viruses in oysters. Nat. Life Sci. Commun. 22(2): e2023034.
INTRODUCTION
Foodborne viruses are predominantly transmitted to humans via fresh and minimally processed foods. They are present in low amounts yet threaten human health. Therefore, sensitive and specific methods are required to monitor viral contaminations in food. Due to their high sensitivity, nucleic acid amplification tests have been used to monitor foodborne viruses. However, molecular viral assays must contain dependable RNA controls or standards to obtain reliable testing. In addition, this type of RNA also serves as a quantitative “standard” or “calibrator” against which the samples are measured. These RNAs are commonly derived from inactivated infectious agents that raise safety concerns. Culturing pathogenic agents is also restricted to specialized laboratories. Alternatively, these RNAs are synthesized from a DNA plasmid by in vitro transcription. Nonetheless, RNA in its naked form is highly vulnerable to RNase, which is ubiquitous in the environment. Using long-term storage of RNA can lead to unreliable detection (Fleige and Pfaffl, 2006).
Naked RNA can be encapsidated to protect from RNase within virus-like particles (VLPs) that are self-assembled from one or several structural proteins. The particles mimic the conformation of a native virus, but without genetic materials. VLPs can be synthesized in cell-free systems, though the production yields are incredibly low (Lingappa et al., 2005). However, VLPs can be obtained in large quantities from a heterologous expression of the corresponding cloned genes. In recent years, VLPs have received noticeable attention to be used as a control for in vitro diagnostics (Beld et al., 2004; Eisler et al., 2004) along with vaccines (Khudyakov, 2008; Roldão et al., 2010; Liu et al., 2012), targeted drug or gene delivery, and nanotechnology (Soto and Ratna, 2010). With a good safety profile, VLPs were suitable as a nanoparticle to protect RNA (Fang et al., 2018). Simple bacteriophages, MS2 and Qβ, have mainly gained attention from researchers as they infect Escherichia coli. Their RNA genome can be encapsidated within their particles formed by 180 copies of capsid protein (CP) with diameters of roughly 28-30 nm (Golmohammadi et al., 1996; Machida and Imataka, 2015). The self-assembly of VLPs can be initiated by co-expressing recombinant RNAs and CPs containing Qβ hairpin (hp) from separately cloned plasmids in E. coli. VLPs were developed for applications in diagnostic procedures. RNAs encapsidated within MS2 VLPs are now used as controls or standards for the detection of various viruses, such as human immunodeficiency virus (Pasloske et al., 1998; Zhan et al., 2009), severe acute respiratory syndrome coronavirus (Drosten et al., 2001), enteroviruses (Beld et al., 2004; Donia et al., 2005), Avian influenza virus (Das et al., 2006), measles virus (D. Zhang et al., 2015; L. Zhang et al., 2015), and ebola virus (Wang et al., 2015). Fang and co-workers (Fang et al., 2017) previously demonstrated the packaging of non-viral RNA in Qβ VLPs. Notwithstanding, the synthesized VLPs required ultracentrifugation in CsCl or sucrose-gradients to purify VLPs, and therefore, they cannot be implemented in laboratories with essential equipment.
In this study, we aimed to produce RNA-based standard reagents for HAV or norovirus detections which rely on VLP technology. The study was also conducted to optimize the final VLP yields. Enclosed RNA was also assessed for use as the standard reagent in oyster sample. Unlike amid reverse transcription (RT)-polymerase chain reaction (PCR), RT-recombinase polymerase amplification (RPA) assay was unaffected by inhibitors found in food and carried out only with fundamental apparatus to maintain a constant temperature. For these reasons, we chose a two-step RT-RPA assay to detect RNA extracted from VLPs and asses for use as a standard for application in clinical laboratory tests and diagnostics.
MATERIALS AND METHODS
Samples
Fresh oysters from local markets were thoroughly cleaned by scrubbing their outer surface and rinsing with water. The whole forms were pooled into a 2-g individual sample and homogenized for nucleic-based analysis.
Primer designs
In this study, all primers were designed to specifically bind the conserved region of the coat protein gene, VP1 of all HAV, and norovirus strains known to date. Primers with a length of 35-bp were synthesized (Macrogen, Seoul, South Korea) and screened for amplification efficiency on agarose gel electrophoresis. The primer pairs of HAV and norovirus (Table 1) amplify 203- and 232-bp products from the former and the latter. Their specificity was also assessed against non-target enteric viruses.
Table 1. Primer sets used in this study.
Construction of Qβ coat protein (CP) and RNA expression plasmids
|
Oligo name Oligo sequence (5' to 3') Tm (°C) |
||
|
Amplification reactions |
||
|
HAV_F4 |
TCTACTGAGCAGAATGTTCCTGATCCCCAAGTCGG |
76.9 |
|
HAV_R1 |
CTGATGTATGTCTAAACTCTCCAGGTTTCAATTCA |
70.9 |
|
NOV_F2 |
GCACCTGTAGCGGGCCAACAAAATGTAATTGACCC |
76.9 |
|
NOV_R2 |
CTGTGAACGCGTTCCTCACTAGAATTACCTGCACT |
74.9 |
|
VLP construction |
|
|
|
Qβ_CP F |
AGTTCCATGGATGGCTAA |
51.4 |
|
Qβ_CP_R |
AACTCCTAGGTCAGTGAT |
51.4 |
|
VLP F |
AGTTTCTAGAAATATATA |
40.3 |
|
VLP R |
AACTGCTAAGCATGCTGT |
51.4 |
A two-plasmid expression system was used to produce VLPs (Figure 1). pCDF-CP was initially generated by amplifying synthetic DNA encoding Qβ CP monomer (NC_001890) including via PCR with the Qβ CP primer pair (Table 1). The resultant DNA fragment including 3' hexahistidine (6X) Tag was digested with NcoI and AvrII and cloned into pCDF-1b vector (Addgene, Watertown, Massachusetts, USA). Transformants were selected on Luria agar plates (LA) containing 50 µg/mL streptomycin (Sm). The RNA expression plasmids, pET-HAV, and pET-NoV, were also produced through PCR using primers specifically bound to 5'spacer region and 3'Qβ hairpin of synthetic DNA (Table 1). Amplified fragments of 422 bp and 352 bp containing the VP1 gene of HAV (Accession No. EF207320) and norovirus (Accession No. MG78678), respectively were cloned at 5'XbaI and 3'BlpI restriction sites into pET-28b(+) (SnapGene, San Diego, California, USA). Transformants growing on an LA plate with 50 µg/mL kanamycin (KAN) were selected. Prior to expression, all transformants were screened for insert identity through double-enzyme digestion, PCR, and DNA sequencing.
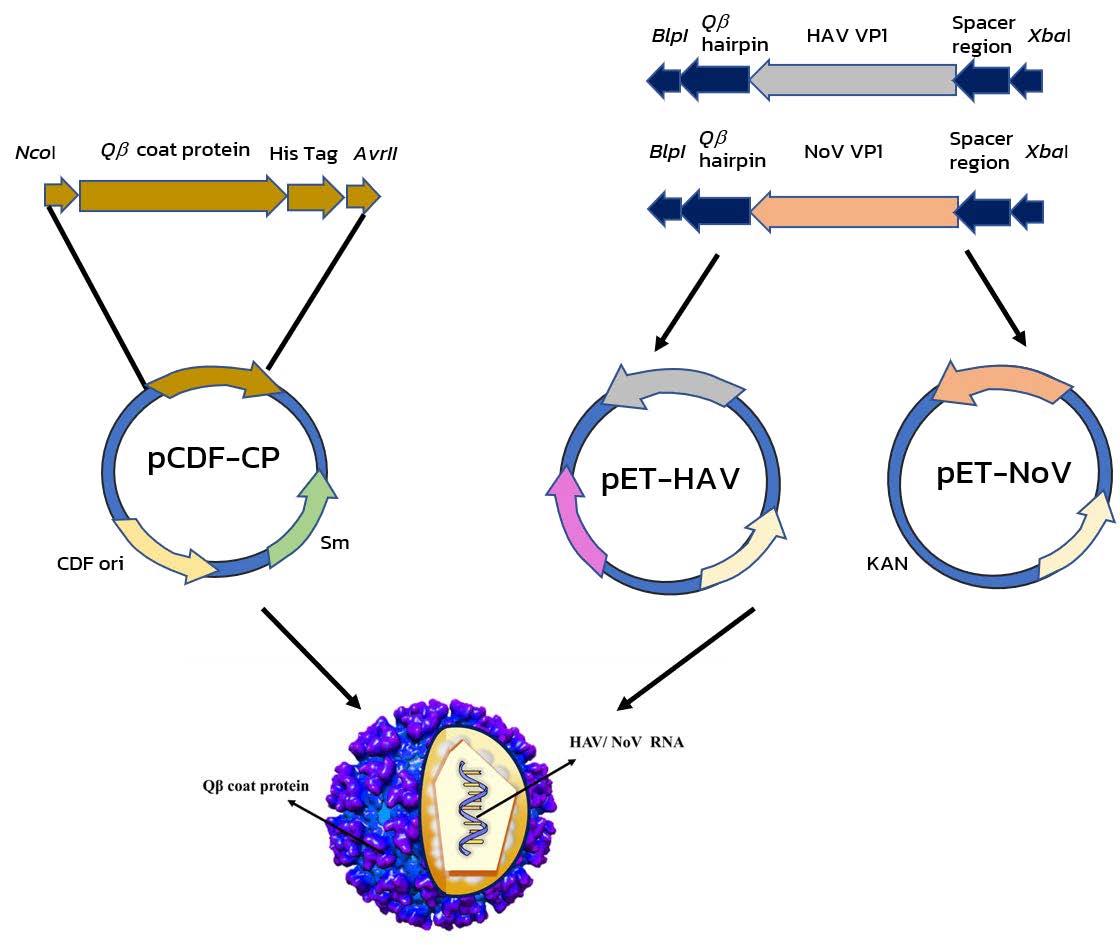
Figure 1. Schematic Flow diagram of methods used to construct the hepatitis virus- and norovirus viral-like particles (VLPs).
Expression of Qβ CP and viral RNA
The recombinant plasmids were expressed under T7 promoter/lac operon control in E. coli BL21 (DE3). pCDF-CP was co-transformed with either pET-NoV or pET-HAV into E. coli host and selected on 50 µg/mL Sm and KAN. A group of single colonies were inoculated into 5 mL NYZ medium [1% selected peptone (N.Z. amine)], 0.5% sodium chloride, 0.5% yeast extract) with the corresponding antibiotics. After being incubated overnight at 37°C Incubator (Contherm, Wellington, New Zealand), one milliliter of the bacterial culture was transferred into 100 mL of ZYM-5052 auto-induction medium (Studier, 2005) and allowed to continue growing for 24 h. After centrifuging at 6,500g, 4ºC for 30 min, the cell pellets were collected and resuspended in an equal volume of Qβ buffer (10 mM MgCl2, and 20 mM Tris-HCl, pH 7.5). Then 10 µL of 10X protease inhibitor was added to the cell suspension in the final 1%. The VLPs were released from host cells by sonication (Sonicator Probe: UP50H (Hielscher, Teltow, Germany) with 10-s on/off intervals on ice for 10 min or until the crude lysates were clear. The crude lysates were adjusted to pH between 3 and 5. Non-pH adjusted (pH 6) and pH-adjusted lysates were centrifuged (KITMAN-T24, TOMY, Tokyo, Japan) at 1600g for 5 min. After discarding cell debris, the supernatant containing RNA enclosed VLPs was collected. Empty Qβ VLP extracted from cells harboring only pCDF-CP was used as a control.
Protein purification by nickel -nitrilotriacetic acid (Ni-NTA) chromatography
VLPs in the crude lysates were isolated using Ni-NTA chromatography (HisPur™ Ni-NTA Chromatography Cartridge, Thermo Scientific, Waltham, Massachusetts, USA) based on the 6XHis Tag design at the carboxyl-terminal of Qβ CPs. The cartridge was equilibrated with 5–10 column volumes of an equilibration/binding buffer (20 mM sodium phosphate, 300 mM sodium chloride, and 10 mM imidazole at pH 7.4) at a flow rate of 1–2 mL/minute for 1-mL cartridge. Bacterial lysate (10 mL) was mixed with the equilibration/binding buffer at a ratio of 1:1 and applied to the cartridge. Subsequent to washing the resin with 10–15 column volumes of a wash buffer (20 mM sodium phosphate, 300 mM sodium chloride, and 20–40 mM imidazole at pH 7.4), the coat proteins of VLPs were eluted with an elution buffer (20 mM sodium phosphate, 300 mM sodium chloride, and 300 mM imidazole at pH 7.4). Purified VLPs (2 mL) were assessed for the protein concentrations using a Pierce BCA protein assay kit (Thermo Scientific, Waltham, Massachusetts, USA) and compared with a bovine serum albumin standard curve. The proteins were stored at −80°C until ready to be employed amid experimentations.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analyses
SDS-PAGE was utilized to analyze the molecular weight of Qβ CP monomers with 12% separating gel compared with the Precision Plus Protein Dual Color Standard protein marker (Bio-Rad, Hercules, California, USA). The protein gels were stained resultant of continuous shaking in solution (0.1% (w/v) Coomassie blue R-250 (CBB), 20% (v/v) methanol, 10% (v/v) acetic acid) for 30 min and destained in solution (50% (v/v) methanol and 10% (v/v) acetic acid) until the gel background was clear. The protein bands on a replica gel were transferred onto a nitrocellulose membrane in transfer buffer (192 mM glycine, 25 mM Tris 10% (v/v) methanol) at 25 V overnight. The membranes were soaked in blocking solution for 4 h at room temperature. The anti-Histidine tag, which was the primary antibody (1:2000), was added and incubated for 2 h at room temperature. Post-washing, the protein complexes were bound with the secondary antibody (optimized horseradish peroxidase reagent working dilution 1:5000) for 1 h and detected with KPL 1-component TMB Membrane Peroxidase substrate (Gaithersburg, Massachusetts, USA) until protein bands of the expected 14.5 kDa were visualized.
Transmission electron microscopy
Ten microliters of samples were deposited on a formvar film-coated copper grid and incubated for 2 min. After drying on a grid for 12 h, the grid was stained in 15 µL of 2% aqueous uranyl acetate for 2 min. The dried grids were kept at 25°C prior to imaging with a Phillips Tecni20 transmission electron microscope.
Preparation of RNA transcribed in vitro
A total of 20 µl reaction contains 2 µL of T7 RNA Polymerase Mix, 10 µL of NTP buffer, and 1 µg of plasmids in 8 µL of dH2O. After incubation at 37°C for 4 h, DNase I was added to the reaction to eliminate the plasmid DNA. The transcribed RNA products were purified prior to analysis for quantity and quality by a nanodrop (ND-2800-ODJ Nano DOT Nucleic Acid Analyzer, Hercuvan Lab system, Malaysia) and denaturing RNA electrophoresis. All RNAs were aliquoted and stored at -80°C until use.
Preparation of denaturing gel electrophoresis for RNA analysis
Eight microliters of RNA transcribed product were first mixed with 2 µL of 10X MOPS Buffer (200 mM MOPS, 50 mM sodium acetate, 10 mM EDTA∙2H2O, 10 mM EGTA). The 10 µL formamide was then added to the mixture to obtain a total of 20 µL mixture. The mixtures were heated at 70°C for 10 min. After suddenly chilling with ice for ≥1 min, the RNA mixtures were added with 2 µL formaldehyde loading buffer (1 mM EDTA, pH 8.0, 0.4% bromophenol blue, xylene cyanol, and 50% glycerol). The RNAs were analyzed in 1% agarose gel electrophoresis containing 1X MOPS Buffer and 37% (v/v) formaldehyde at 50 V for 60 min. The RNA bands were visualized with 1 µg/mL ethidium bromide.
RNA extraction from VLPs
Fifty micrograms of viral VLPs were resuspended in 250 µL of extraction buffer (5% SDS, 25 mM DTT, 20 mM Tris-HCl, pH 7.5, and 50 mM NaCl). VLP suspension was boiled to release RNA at 70°C for 5 min or extracted with the TrizolTM reagent (Thermo Scientific, Waltham, Massachusetts, USA) following the manufacturer’s instructions. In spiking experiments, 2 g of virus-free oysters were added with 5 µL of 10-fold diluted of HAV-VLPs or norovirus-VLPs and incubated at 37°C for 1 h. Each spiked sample was then added with 2 mL of 100 µg/mL proteinase K (Biotechrabbit, Hennigsdorf Germany). The mixtures were incubated at 37°C for 1 h and then at 60°C for 15 min under continuous agitation. The cell debris was separated via centrifugation at 14,000g for 5 min and filtered twice with filter papers. To obtain RNA, the flow-through was either boiled at 70°C for 5 min or extracted with the TrizolTM reagent (Thermo Scientific, Waltham, Massachusetts, USA). Nanodrop and agarose gel electrophoresis were used to check RNA integrity and concentration. RNAs were then applied to amplify cDNAs by RT.
Reverse transcription (RT)
The RNAs were reverse transcribed to obtain the single-stranded cDNA using a Random Hexamer RevertAid First Strand cDNA Synthesis kit (Thermo Scientific, Waltham, Massachusetts, USA). Following the manufacturer’s instructions, a total volume of 20 µL reaction contains the total RNAs (0.1 ng–5 µg), 1 µL of oligo dT Random Hexamer primer, 4 µL of 5X Reaction Buffer, 1 µL of Ribolock RNase inhibitor (20 U/µL), 2 µL of 10 mM dNTPs mix, and 1 µL of RevertAid M-MuLV RT (200 U/µL). The mixture was initially incubated at 25°C for 5 min, followed by 42°C for 60 min. The reaction was stopped by shifting the temperature to 70°C for 5 min. The resulting single-stranded cDNAs were used as the templates for the downstream RPA reaction. The copy number was calculated according to the following Equation.
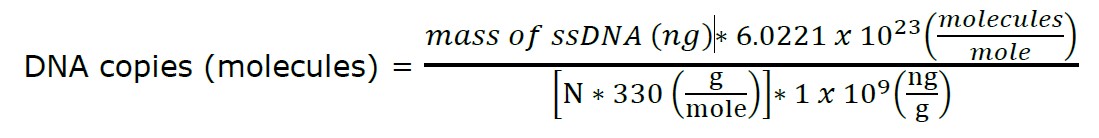
Recombinase polymerase amplification (RPA)
Following the manufacturer’s instructions (TwistAmp Ltd., Cambridge, UK), 1 µL of cDNA templates was added to 29.5 µL TwistAmp rehydrate buffer containing 0.42 µM forward and reverse primers. Two and a half microliters of 14 mM Mg (Ac)2 were then added to start the 50 µL of reaction at 37°C for 20 min. Subsequent to purification using a GenepHlowTM Gel/PCR Kit (Geneaid, New Taipei City, Taiwan), the purified amplification products were analyzed on 1% (w/v) agarose gels containing 1X SYBR Safe (Invitrogen by Thermo Fisher Scientific, USA) in 1X Tris-Boric acid-EDTA (TBE) buffer.
Assessment of enclosed RNAs used as a standard
Serial 10-fold diluted cDNA templates were made. One microliter of each diluted template was employed in RPA reaction, and their amplified products were analyzed through agarose gel electrophoresis. Band intensities of these products on agarose gel were measured using the Image J program (developed by the National Institutes of Health and Laboratory for Optical and Computational instrumentation). Data were subsequently converted to molecules. In addition, RNA products from in vitro transcription were also applied as templates in RT-RPA. After gel electrophoresis, a graph was generated. Band intensity of the reaction products of RNA from VLP was set on the Y-axis and that of transcribed RNA on the X-axis. In this study, each dilution was tested in duplicate, and the whole assay was performed twice.
Statistical analysis
Standard curves were generated and linear regression was used to test for equivalence of of RNAs enclosed VLPs versus naked RNAs. Data with a correlation co-efficiency of r2 ≥ 0.9 was considered to be acceptable for analysis.
DNA verification by sequencing
Amplified RT-RPA products were purified from the agarose gel (GeneHlow Gel/PCR kit; Geneaid, New Taipei City, Taiwan). DNA sequencing was used to verify their sequence identities (Macrogen, Seoul, South Korea).
RESULTS
Expression of DNA plasmids and characterization of QβVLPs
Expression plasmids were tested for their expression ability by in vitro transcription. The resulting RNA were visualized on 1% denaturing agarose gel electrophoresis (data not shown). After in vivo assembly via co-expressing recombinant RNAs and CPs, the VLPs were extracted from E. coli BL21(DE3) through sonication and purified using Ni-NTA chromatography. SDS-PAGE and western blot analysis showed that pCDF-CP alone, or either with pET- HAV or pET- NoV expressed the CP monomer (Figure 2A and 2B, respectively). All purified constructs showed a distinguished CPs protein band with a molecular weight of 14.5 kDa without proteolytic cleavage. To ensure that the constructed VLPs remain intact, the morphology of VLPs was visualized via transmission electron microscopy. Low with variably-sized particles were observed (Figure 2C).
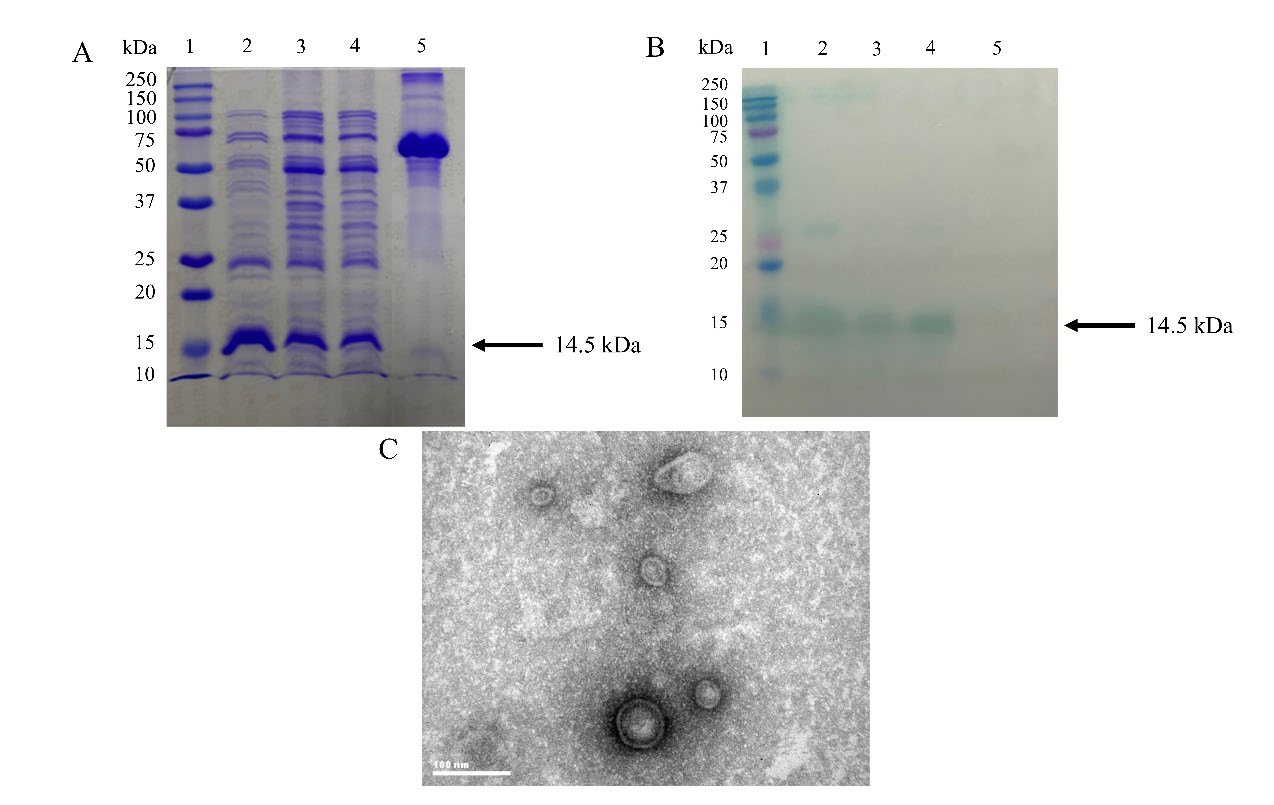
Figure 2. Characterization of viral-like particles (VLPs). (A) SDS-PAGE and (B) Western blot using anti-His Tag antibody. Lane 1: The Precision Plus Protein Dual Color Standard protein marker (Bio Rad, Hercules, California, USA). Lane 2: Qβ coat protein (CP) monomer. Lane 3: HAV-VLPs. Lane 4: NoV-VLPs. Lane 5: Bovine serum albumin was used as the negative control. The arrows on the right of Figures 2A and 2B indicate the expected molecular weight of Qβ CP monomer of 14.5 kDa. (C) Transmission electron microscope images of HAV-VLPs. Scale bar=100 nm.
The production of RNA enclosed VLPs
Following sonication, the crude lysates were adjusted to a pH between 3 and 5. When adjusting pH to lower than 6, SDS-PAGE and western blot displayed a more intense protein band than at pH 6 (Figure 3A and 3B, respectively). We selected crude lysates at pH 4 which seems to be the optimal condition for protein expression to finally prove the whole icosahedral formation of the VLPs. Transmission electron microscopy revealed substantial particles with homogenous particle size at an average diameter of 30 nm (Figure 3C). To ensure the packaging of RNA in particles, VLPs were heated at 70°C for 5 min. Under such conditions, RNA can be quickly released from its packaging by which its yield is roughly comparable to that of the TrizolTM reagent (Figure 4). The production yield of HAV and norovirus packaged RNA is about 68.89 ng and 126 ng of RNA for every 50 ug VLP (protein) measured through spectrophotometry. The Equation can convert these RNA contents to 1.2x107 copies/ng of the former and 1.9x107 copies/ng of the latter.
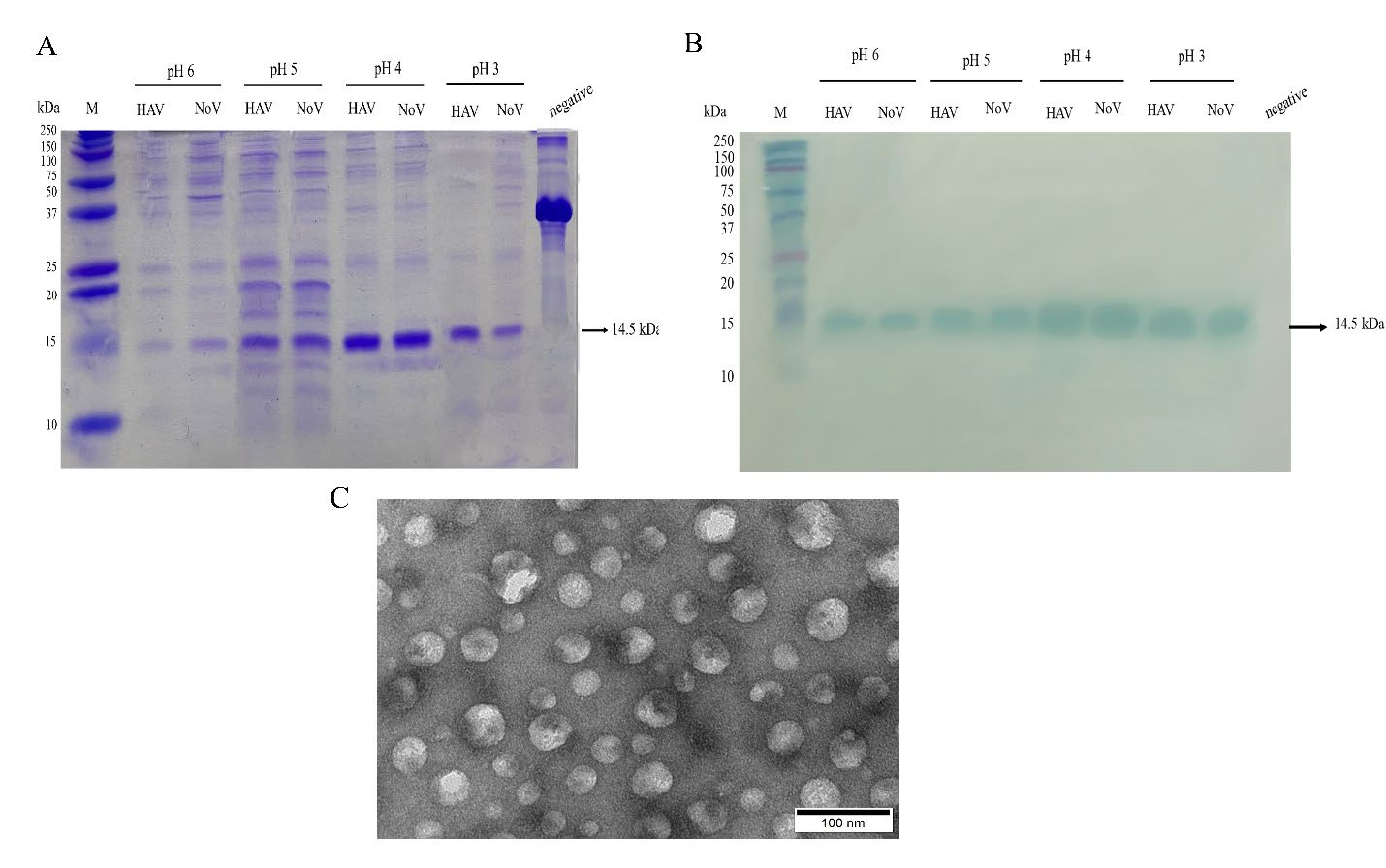
Figure 3. The effect of pH on the production of VLPs. (A) HAV-VLP and NoV-VLPs in the lysates at pH 3 to 6 were characterized by SDS-PAGE and (B) Western blot using anti-His Tag antibody. M: The Precision Plus Protein Dual Color Standard protein marker (Bio Rad, Hercules, California, USA). Bovine serum albumin was used as the negative control. The arrows on the right of Figures 2A and 2B indicate the expected molecular weight of Qβ CP monomer of 14.5 kDa. (C). Transmission electron microscope images of HAV-VLPs from the lysate at pH 4. Scale bar=100 nm.
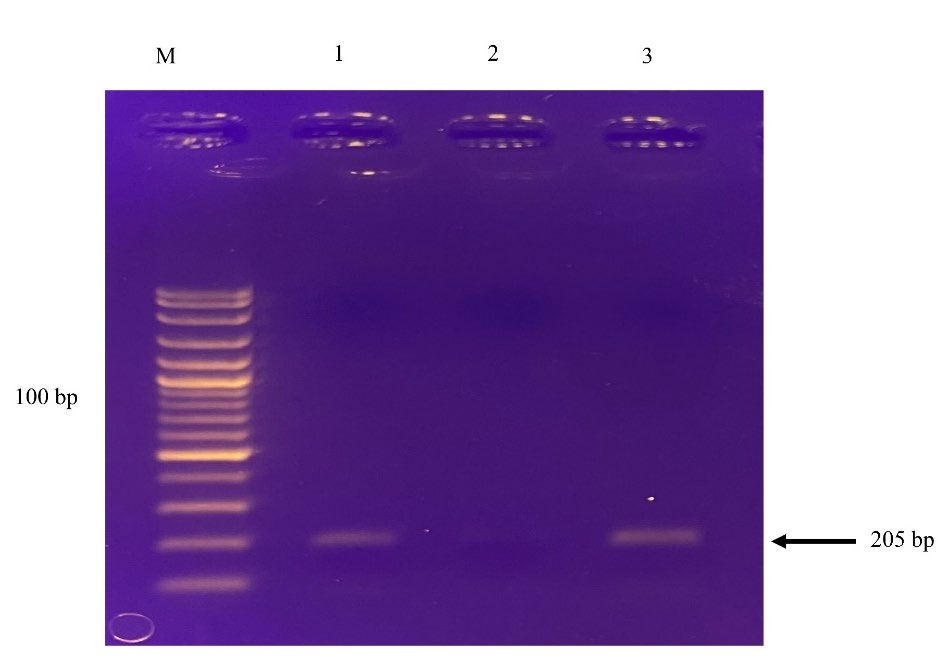
Figure 4. The packaging of RNA in VLPs. RNAs were extracted from VLPs and compared with TrizolTM reagent. RNA samples were used as templates in the two-step RT-RPA. The amplified products from heated VLPs (Lane 1), unheated VLPs (Lane 2), and TrizolTM reagent (Lane 3) were visualized on 1% agarose gel electrophoresis.
Assessment of VLPs used as a standard
RT-RPA yielded a clear signal when RNA extracted from purified VLPs was at a minimum of 4.54×104 molecules. In the spiking experiment, a minimum of RNA was detected by RT-RPA at 1.34×1010 molecules. To study the relationship between transcribed RNA and RNA extracted from VLP applied as templates of RT-RPA, the R2 value in the linear regression model showed excellent linearity (R2 = 0.9333) (Figure 5).
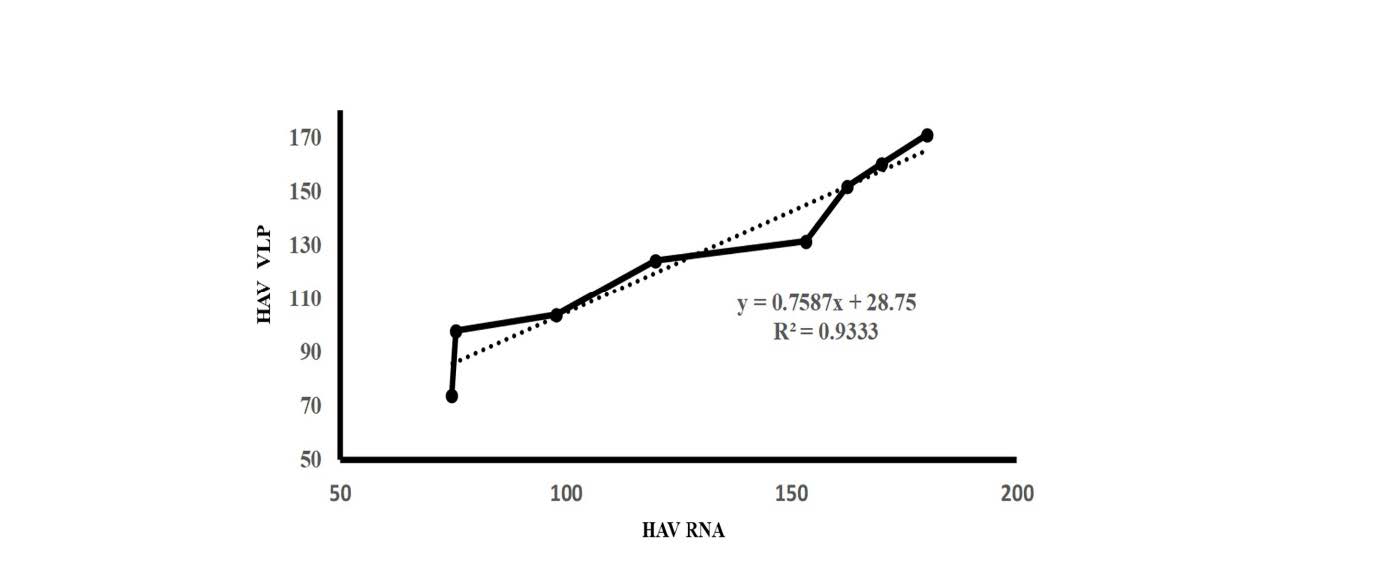
Figure 5. The equivalence of RNA enclosed viral-like particles to naked RNAs. Ten-fold serial dilutions of transcribed RNA and RNA extracted from HAV-VLPs were amplified by RT-RPA. Their band intensities of amplified products of RT-RPA on agarose gel were measured by program J (developed by the National Institutes of Health and Laboratory for Optical and Computational instrumentation). Linear Regress was used to test for the equivalence with R2 = 0.9333.
DISCUSSION
In this study, we proposed a simple and economical method for synthesizing RNA-based standard reagents for HAV or norovirus detections via RT-RPA. Most molecular testing depends on using RNA synthesized by in vitro transcription for positive controls and external standards. Although transcribed RNAs are easier to prepare for detection, long-term storage affects their stability, thus leading to unreliable testing. Based on VLP technology, RNA was enclosed in a viral protein shell composed of Qβ CP monomers which are self-assembled in their precise and repeated structures. The thermodynamic stability of the icosahedral structure of VLPs protects enclosed RNA against RNases (WalkerPeach et al., 1999) and small diffusible species (Fang et al., 2018). In addition, RNA is more compact in the relatively large cavity space of VLPs and less dynamic than RNAs free in solution (Dent et al., 2013), thus making it less vulnerable to RNases.
Native Qβ bacteriophages lyse E. coli host and release particles into the culture media. However, synthesized VLPs were localized in the cytoplasmic fraction of E. coli and required disruption of cells by sonication. During a purification process, the extraction buffers are supplemented with reducing and chelating agents and protease inhibitors to protect the VLPs from oxidation and host proteases. After clarification through low-speed centrifugation, the VLP-containing solutions have to be concentrated by filtering to decrease the volume of the extract. Further VLP purification reduces the amount of host-derived impurities. Instead of using ultracentrifugation in CsCl or sucrose gradients, adding 6XHis-Tag at the carboxyl-terminal of Qβ CPs facilitates purification through affinity chromatography. The Tag design also aids in identifying the VLP structural proteins with western blot analysis using an anti-His tag antibody. However, amino acid substitutions could interfere with CP folding and result in VLP instability (Stonehouse and Stockley, 1993). In this study, the introduced Tag directly into the VLP structure did not lead to proteolytic cleavage as seen as a distinct band of 14.5 kDa on the western blot. Our result agreed with a previous report of including the His-tag at the N-terminal part of the CP of potato virus Y-like particles baculovirus (Kalnciema et al., 2012). For complete characterization, an electron microscope analysis is necessary as the visual proof of the newly synthesized VLPs. In our study, the added Tag did not interfere with the whole icosahedral formation of the VLPs. These particle sizes of about 30 nm were observed. Nevertheless, the yield of VLP production was low with variably-sized particles, which probably resulted from large, unstructured protein aggregates.
Several physicochemical factors can affect VLP stability; pH is one such factor. Some studies demonstrated that bacteriophage MS2 could stay stable at pH 7 despite becoming less stable as pH decreases (Lago et al., 2001). In this study, when adjusting the pH of the lysate after sonication to lower than 6, the increased abundance of CP bands was visible on the western blot. The production yield of QβVLPs also improved at pH 4 probably due to the fact that, at a lower pH, non-VLP particles and aggregates were removed from filtration. Thus, the filtered solution substantially retains the complete and highly uniform VLP particles, as clearly seen with the EM. Besides pH factors, temperature also strongly impacts the stability of VLPs. Stonehouse and Stockley (Stonehouse and Stockley, 1993) found that the MS2 phage had a melting point of 66°C and can withstand temperatures as high as 68°C whereas Qβ VLPs are stable over a broad range of temperatures (Fiedler et al., 2010). In addition to those factors, the stability of VLPs can also be markedly raised by introducing disulfide bridges to crosslink the individual CPs (Bundy and Swartz, 2011).
Heating at 70°C for 5 min quickly released the packaged RNAs from the Qβ CP. The yield of RNAs was roughly comparable to that extracted from the TrizolTM reagent. In our study, the production yield of HAV and norovirus RNA was 68.89 ng and 126 ng for every 50 ug VLP (protein) measured by spectrophotometry or 1.2x107 copies/ng and 1.9x107 copies/ng, respectively. Sufficient RNA was packaged in the particles to fully compensate for a cationic charge of Qβ CP (Muriaux et al., 2001). The packaging efficiency of our results was consistent with that in a plant cowpea mosaic virus (CPMV) in the range of 102-108 copies/ ng of VLP (Peyret et al., 2022). Still, it was lower than what was previously reported (5 µg) (Fang et al., 2018). The differences in packaging efficiency possibly resulted from the resistance markers of the expression plasmids and the composition of the cultivation medium that influenced the assembly of Qβ VLPs (Brown et al., 2009). The efficient packaging of RNA within VLPs in vivo may also depend upon the compactness of the target RNA structure, the absence of competing for binding motifs in the RNA, besides the presence of the Qβ hp (Fang et al., 2018). In this study, HAV, and norovirus RNAs presented a similar length of approximately 200 nucleotides. They could be encapsidated within VLPs with equal packaging efficiency. Notwithstanding, RNA length is not a solid predictor of packaging efficiency, though intrinsic RNA compaction affects the effectiveness of RNA wrapping within Qβ VLPs (Fang et al., 2018). Even in the same type of VLPs, we cannot assume that every particle is filled with the same amount of RNA or devoid of any RNA. If needed, density gradient ultracentrifugation can be used to distinguish Qβ VLPs based on their RNA contents. Since virus detection via RT-RPA in the food matrix is not anticipated, this was not performed for the current investigation.
Since the recombinant RNA in VLPs can be released from its packaging by low-temperature heating, VLPs can be added directly to the oyster samples in the spiking experiment. Our results revealed that both VLP-packaged and free recombinant RNA from in vitro transcription presented no difference in detection via RT-RPA. Accordingly, these results could arise from using newly synthesized RNA rather than RNA kept for an extended period. In addition, linear regression analysis strongly emphasizes the alternative use of encapsidated RNA to naked RNA.
The plasmid, a highly stable form of nucleic acid, can be transcribed into RNA through in vitro transcription. Although this method is effective, it can be time-consuming and expensive. In theory, RNA products from this reaction can be stored for extended periods at a temperature of -80°C. However, our observations, which were made using agarose gel electrophoresis and spectrophotometry detection, revealed that RNA samples from in vitro transcription tend to gradually degrade over time, with a degradation rate of approximately 25% after one week. This phenomenon may be attributed to fluctuations in temperature within the deep freeze. While freezers set to -20°C are more commonly found in laboratories with basic equipment, they are only suitable for short-term storage of RNA. Our experience indicates that RNA extracted from VLPs stored at -20°C for 15 months still exhibited visible results on an agarose gel, and hence, better amplifiability. In addition, it should be noted here that the utilization of HAV and norovirus Qβ VLPs presents numerous advantages over that of an intact virus. Production of a human virus relies on a cell culture system which is time-consuming. Culturing HAV and human norovirus can be even more challenging due to the lack of a cultivation system. Furthermore, norovirus has a high mutation rate, resulting in a non-homogenous sequence of a standard. Recombinant RNAs thus provide a well-characterized sequence less likely to contain variations.
CONCLUSION
VLP preparation methods are designed to be suitable for RNA storage in basic or minimally equipped laboratories. These encapsidated RNAs can be used as the working standards in detecting foodborne viruses. Moreover, the high yield of these VLP particles and stability of the RNAs allow for cost-effective production. Hence, this construct may become an ideal “control” and “standard” for application in clinical laboratory tests and diagnostics.
ACKNOWLEDGEMENTS
We are grateful to Professor Dr. Somsak Pantuwatana, and Professor Dr.Wanpen Chaicumpa, Faculty of Medicine, Siriraj Hospital, Mahidol University, for valuable advice and great support.
AUTHOR CONTRIBUTIONS
Uraiwan Intamaso designed the experiments and wrote the manuscript. Palatip Chutoam performed the statistical analysis and data visualization. Supannee Lethochavalit collected specimen. Suthasinee Jinda conducted the experiments. All authors have read and approved of the final manuscript.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
Beld, M., Minnaar, R., Weel, J., Sol, C., Damen, M., van der Avoort, H., Wertheim-van Dillen, P., van Breda, A., and Boom, R. 2004. Highly sensitive assay for detection of enterovirus in clinical specimens by reverse transcription-PCR with an armored RNA internal control. Journal of Clinical Microbiology. 42: 3059–3064.
Brown, S.D., Fiedler, J.D., and Finn, M.G. 2009. Assembly of hybrid bacteriophage Qβ virus-like particles. Biochemistry. 48: 11155–11157.
Bundy, B.C. and Swartz, J.R. 2011. Efficient disulfide bond formation in virus-like particles. Journal of Biotechnology. 154: 230–239.
Das, A., Spackman, E., Senne, D., Pedersen, J., and Suarez, D.L. 2006. Development of an internal positive control for rapid diagnosis of avian influenza virus infections by real-time reverse transcription-PCR with lyophilized reagents. Journal of Clinical Microbiology. 44: 3065–3073.
Dent, K.C., Thompson, R., Barker, A.M., Hiscox, J.A., Barr, J.N., Stockley, P.G., and Ranson, N.A. 2013. The asymmetric structure of an icosahedral virus bound to its receptor suggests a mechanism for genome release. Structure. 21: 1225–1234.
Donia, D., Divizia, M., and Pana’, A. 2005. Use of armored RNA as a standard to construct a calibration curve for real-time RT-PCR. Journal of Virological Methods. 126: 157–163.
Drosten, C., Seifried, E., and Roth, W.K. 2001. TaqMan 5′-nuclease human immunodeficiency virus type 1 PCR assay with phage-packaged competitive internal control for high-throughput blood donor screening. Journal of Clinical Microbiology. 39: 4302–4308.
Eisler, D.L., McNabb, A., Jorgensen, D.R., and Isaac-Renton, J.L. 2004. Use of an internal positive control in a multiplex reverse transcription-PCR to detect West Nile virus RNA in mosquito pools. Journal of Clinical Microbiology. 42: 841–843.
Fang, P.-Y., Bowman, J.C., Gómez Ramos, L.M., Hsiao, C., and Williams, L.D. 2018. RNA: Packaged and protected by VLPs. RSC Advances. 8: 21399–21406.
Fang, P.-Y., Gómez Ramos, L.M., Holguin, S.Y., Hsiao, C., Bowman, J.C., Yang, H.-W., and Williams, L.D. 2017. Functional RNAs: Combined assembly and packaging in VLPs. Nucleic Acids Research. 45: 3519–3527.
Fiedler, J.D., Brown, S.D., Lau, J., and Finn, M.G. 2010. RNA-directed packaging of enzymes within virus-like particles. Angewandte Chemie (International Edition). 49: 9648–9651.
Fleige, S., and Pfaffl, M.W. 2006. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27: 126–139.
Golmohammadi, R., Fridborg, K., Bundule, M., Valegård, K., and Liljas, L. 1996. The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure (London, England: 1993). 4: 543–554.
Kalnciema, I., Skrastina, D., Ose, V., Pumpens, P., and Zeltins, A. 2012. Potato virus Y-like particles as a new carrier for the presentation of foreign protein stretches. Molecular Biotechnology. 52: 129–139.
Khudyakov, Y.E. (Ed.). 2008. Construction of novel vaccines on the basis of virus-like particles: Hepatitis B virus proteins as vaccine carriers. In Medicinal Protein Engineering. CRC Press.
Lago, H., Parrott, A.M., Moss, T., Stonehouse, N.J., and Stockley, P.G. 2001. Probing the kinetics of formation of the bacteriophage MS2 translational operator complex: Identification of a protein conformer unable to bind RNA11. Journal of Molecular Biology. 305: 1131–1144.
Lingappa, J.R., Newman, M.A., Klein, K.C., and Dooher, J.E. 2005. Comparing capsid assembly of primate lentiviruses and hepatitis B virus using cell-free systems. Virology. 333: 114–123.
Liu, F., Ge, S., Li, L., Wu, X., Liu, Z., and Wang, Z. 2012. Virus-like particles: Potential veterinary vaccine immunogens. Research in Veterinary Science. 93: 553–559.
Machida, K. and Imataka, H. 2015. Production methods for viral particles. Biotechnology Letters. 37: 753–760.
Muriaux, D., Mirro, J., Harvin, D., and Rein, A. 2001. RNA is a structural element in retrovirus particles. Proceedings of the National Academy of Sciences of the United States of America. 98: 5246–5251.
Pasloske, B.L., Walkerpeach, C.R., Obermoeller, R.D., Winkler, M., and DuBois, D.B. 1998. Armored RNA technology for production of ribonuclease-resistant viral RNA controls and standards. Journal of Clinical Microbiology. 36: 3590–3594.
Peyret, H., Groppelli, E., Clark, D., Eckersley, N., Planche, T., Ma, J., and Lomonossoff, G.P. 2022. Production and use of encapsidated RNA mimics as positive control reagents for SARS-CoV-2 RT-qPCR diagnostics. Journal of Virological Methods. 300, 114372.
Roldão, A., Mellado, M.C.M., Castilho, L.R., Carrondo, M.J.T., and Alves, P.M. 2010. Virus-like particles in vaccine development. Expert Review of Vaccines. 9: 1149–1176.
Soto, C.M. and Ratna, B.R. 2010. Virus hybrids as nanomaterials for biotechnology. Current Opinion in Biotechnology, 21: 426–438.
Stonehouse, N.J. and Stockley, P.G. 1993. Effects of amino acid substitution on the thermal stability of MS2 capsids lacking genomic RNA. FEBS Letters. 334: 355–359.
Studier, F.W. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification. 41: 207–234.
WalkerPeach, C.R., Winkler, M., DuBois, D.B., and Pasloske, B.L. 1999. Ribonuclease-resistant RNA controls (armored RNA) for reverse transcription-PCR, branched DNA, and genotyping assays for hepatitis C virus. Clinical Chemistry. 45: 2079–2085.
Wang, G., Sun, Y., Zhang, K., Jia, T., Hao, M., Zhang, D., Chang, L., Zhang, L., Zhang, R., Lin, G., Peng, R., and Li, J. 2015. External quality assessment of molecular detection of ebola virus in China. PLoS One. 10: e0132659.
Zhan, S., Li, J., Xu, R., Wang, L., Zhang, K., and Zhang, R. 2009. Armored Long RNA controls or standards for branched DNA assay for detection of human immunodeficiency virus type 1. Journal of Clinical Microbiology. 47: 2571–2576.
Zhang, D., Sun, Y., Jia, T., Zhang, L., Wang, G., Zhang, R., Zhang, K., Lin, G., Xie, J., Wang, L., and Li, J. 2015. External quality assessment for the detection of measles virus by reverse transcription-PCR using armored RNA. PLOS ONE. 10: e0134681.
Zhang, L., Sun, Y., Chang, L., Jia, T., Wang, G., Zhang, R., Zhang, K., and Li, J. 2015. A novel method to produce armored double-stranded DNA by encapsulation of MS2 viral capsids. Applied Microbiology and Biotechnology. 99: 7047–7057.
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand.
Uraiwan Intamaso1, *, Palatip Chutoam1, Suthasinee Jinda1, and Supannee Lethochavalit2
1 Faculty of Allied Health Sciences, Burapha University, 169 Long-Hard Bangsaen Rd., Chonburi 20131, Thailand.
2 Institute of Marine Science, Burapha University, 169 Long-Hard Bangsaen Rd., Chonburi 20131, Thailand.
Corresponding author: Uraiwan Intamaso, E-mail: uraiwani@go.buu.ac.th
Total Article Views
Editor: Veerasak Punyapornwithaya,
Chiang Mai University, Thailand
Article history:
Received: November 11, 2022;
Revised: March 14, 2023;
Accepted: March 21, 2023;
Published online: March 29, 2023