 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Characterization of D-Glucoside 3-Dehydrogenase from Rhizobium sp. L35 and Its Application for D-Allose Production. Akkharapimon Yotsombat, Keiko Uechi, Shunsuke Onishi, Kohei Mino, Tae Hasegawa, Kenji Morimoto, and Goro Takata*
Published Date : 2019-08-23
DOI : https://doi.org/10.12982/CMUJNS.2019.0019
Journal Issues :
Number 3 , July-September 2019
ABSTRACT
D-allose is a rare monosaccharide that possesses some interesting fea- tures for enhancing the antitumor effects of chemoradiotherapy and treatment of cancer, however, efficient production of D-allose with high percent yield has not yet been reported. In this study, we proposed a utilization of D-glucoside 3-dehydrogenase together with chemical reaction to improve the D-allose pro- duction. A D-glucoside 3-dehydrogenase, which regioselectively dehydroge- nates glycosides at the C-3 position to the corresponding 3-ketoglycoside, was newly isolated as Rhizobium sp. L35 and characterized as a flavin adenine dinucleotide-dependent dehydrogenase. Its molecular weight was determined to be 67,000 by SDS-PAGE and 131,000 by size-exclusion chromatography, suggesting that it is a dimeric enzyme. Its optimum pH and temperature with respect to activity were 7.5 and 40°C, respectively. It was stable between pH 6.0 and 11.0, and below 40°C (half-life of 3 h at 40°C and 50 min at 45°C). The en- zyme showed broad substrate specificity towards various glycosides, especially β-1,4-linked disaccharides such as cellobiose and lactose. Finally, D-allose production was performed by a three-step process of enzymatic-dehydrogena- tion, chemical reduction and acid-hydrolysis, using cellobiose as the starting material. The yield of D-allose was estimated to be 30% from cellobiose. This result indicates that D-allose can be produced by this strategy three-fold high- er than the conventional method.
Keywords: D-Glucoside 3-dehydrogenase, 3-Ketoglycoside, Oxidoreductase, Rare sugar, D-Allose
INTRODUCTION
D-Glucoside 3-dehydrogenase (G3DH; EC 1.1.99.13) was firstly discov- ered and characterized in Agrobacterium tumefaciens (Hayano et al., 1967) and later found in other organisms such as Flavobacterium saccharophilum (Takechi et al., 1986; Takeuchi et al., 1988), Agaricus bisporus D649 (Morrison et al., 1999), Halomonas sp. α-15 (Kojima et al., 1999), and Stenotrophomonas malto- philia CCTCC M 204024 (Zhang et al., 2006). G3DH is a flavin adenine di- nucleotide-dependent (FAD-dependent) oxidoreductase that catalyzes the de- hydrogenation of hydroxyl group at the C-3 position of D-glucose and other aldopyranosides to their corresponding 3-ketoglucoside and 3-ketopyranosides (Maeda et al., 2001). When disaccharides are provided as substrates, G3DH would catalyze from the non-reducing end of the glycosides (Bernaerts, 1963).
A number of enzymes producing 3-ketoglycosides have been reported, including pyranose oxidase (EC 1.1.3.10) from Phanerochaete chrysosporium (Giffhorn, 2000) and pyranose dehydrogenase (EC 1.1.99.29) from A. bisporus (Volc et al., 1997; Volc et al., 1998), Macrolepiota rhacodes (Volc et al., 2001), and A. xanthodermus (Kujawa et al., 2007). These oxidoreductases are well known for their application for the sugars bioconversion, and for blood glucose level measurement (Kojima et al., 1999). In addition, the utilization of G3DH from Halomonas sp. α-15 for measurement of 1,5-anhydro-D-glucitol, a clinical marker for diabetes, has an advantage over other oxidoreductases because of its regioselectivity property (Tsugawa et al., 1996).
The reaction products, 3-ketoglycosides are attractive for production of commodity chemicals such as polymers and surfactants (Eltz, 1968) and for pro- duction of rare sugars. Rare sugars are defined as saccharides and their deriva- tives occurring only in a small quantity in nature. D-allose, classified as a rare sugar, has been reported several times for its physiological features and has at- tracted attention as a precursor for other unnatural compounds production (Levin et al., 1995; Kasiganesan et al., 2009) and as an anti-tumor drug (Moyroud et al., 1999; Yamaguchi et al., 2008).
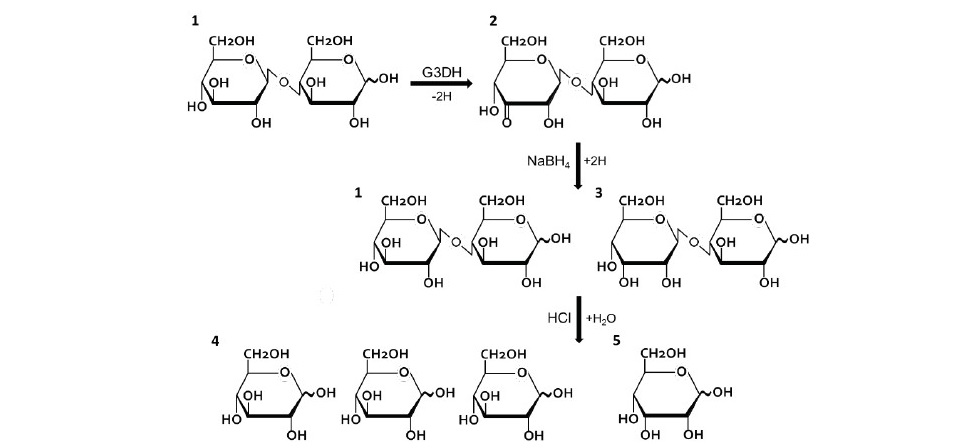
Figure 1. A strategy of D-allose production using G3DH from Rhizobium sp. L35. Cellobiose (β1,4-D-glucosyl-D-glucose; 1) is dehydrogenated to 3-ketocellobiose (β1,4-3-keto-D-glucosyl-D-glucose; 2) by G3DH. Then, 3-Ketocellobiose is equally converted to cellobiose (1) and β1,4-D-allosyl-D-gulcose (3) by chemical reduction using NaBH4. These two products are acid-hydrolyzed to D-glucose (4) and D-al- lose (5), at a theoretical yield of 75% and 25%, respectively.
Mass-production of D-allose has been established based on the Izumoring strategy, using enzymatic conversion of an inexpensive sugar (Granström et al., 2004). D-fructose is epimerized to D-psicose (D-allulose) by D-tagatose 3-epimerase, and then it is isomerized to D-allose by L-rhamnose isomerase with approximately 10% of theoretical yield. We have investigated the utiliza- tion of G3DH isolated from Rhizobium sp. L35 to improve D-allose production. Figure 1 shows a strategy of D-allose production procedure. Cellobiose, as a substrate, is oxidized to 3-ketocellobiose (β-1,4 3-keto-D-glucosyl-D-glucose) by G3DH, and subsequently converted to β-1,4 D-allosyl-D-glucose and cellobiose by chemical reduction under mild conditions. After that, the two di- saccharides are acid-hydrolyzed to D-glucose and D-allose at theoretical yields of 75% and 25%, respectively. Compared with the convention method, G3DH has shown the possibility to achieve mass-production of D-allose. In addition, the production of D-allose by enzymatic reaction would establish a new produc- tion pathway for rare sugar. In this study, G3DH from Rhizobium sp. L35 was purified and characterized to apply for the rare sugar production.
MATERIALS AND METHODS
Organisms, and growth conditions
Several microorganisms were isolated from soil and grown in mineral salt (MS) medium (0.26% (NH4)2SO4, 0.24% KH2PO4, 0.56% K2HPO4, 0.01% MgSO4.7H2O, and 0.05% yeast extract) containing 1% lactitol as a carbon source. Each microorganism was observed for a lactitol oxidation property by the cysteine-carbazole method (Dische and Borenfreund, 1951). One microor- ganism having the highest lactitol oxidation activity was named strain L35 and later identified as Rhizobium sp. by the 16S rDNA sequence analysis (Sanger et al., 1977) using a DTCS Quick Start Kit (Beckman Coulter, Fullerton, CA, USA) and a CEQ8000 system (Beckman Coulter).
To optimize the condition for G3DH production, Rhizobium sp. L35 was cultivated in four types of medium; Mineral salt (MS), Meat Extract (ME; 0.5% meat extract, 0.5% polypeptone, and 0.5% sodium chloride), Yeast Extract (YE; 0.5% polypeptone, 0.5% yeast extract, and 0.5% sodium chloride), and Tryptic Soybean Broth (TSB; 2% tryptic-soybean-broth). All medium was adjusted to pH 7.0. To each medium, 1.0 % of carbohydrate (D-glucose, cellobiose, maltose, trehalose, lactose, sucrose, D-sorbitol, lactitol, maltitol) was added as a carbon source. For standard medium without carbohydrate was used as a control sample.
Enzyme assay and protein assay
G3DH activity was measured by following the reduction rate of 2,6- dichlorophenolindophenol (DCPIP; molar extinction coefficient [ε] under the measuring conditions: ε600 = 10,000 M-1cm-1) at a wavelength of 600 nm using a microspectrophotometer (BioTek Instruments Inc., Winooski, VT, USA). Reaction mixture composed of 5 mM substrate and 20 µM DCPIP in 180 µL of 50 mM Tris-HCl buffer (pH 7.5) was pre-incubated at 37°C for 10 min. The re- action was initiated by adding 20 µL of enzyme into the reaction mixture. This composition was assigned to be a standard condition. One unit (U) of the G3DH activity is defined as an amount of enzyme that causes the reduction of 1 µmol of DCPIP per minute. Protein concentration was measured by the Bradford method (Bradford, 1976) using a Protein Assay Kit (Bio-Rad, Hercules, CA, USA).
Enzyme preparation and purification
Rhizobium sp. L35 was cultivated in 3 L of YE medium containing 1% maltitol at 30°C for 24 h. The cells were harvested by centrifugation at 10,000 rpm for 10 min and washed twice with 50 mL of 20 mM Tris-HCl buffer (pH 7.5). The cell pellet was resuspended in 50 mL of the same buffer containing 1 mM EDTA and 1 mM phenylmethylsulfonyl fluoride (PMSF) before disruption by sonication (Branson Sonifier 450, Danbury, CT, USA) in ice-cooling bath. After extraction, the suspension was centrifuged at 18,000 rpm for 30 min to remove the cell debris and the supernatant was used as a crude extract.
For purification, the crude extract containing G3DH was previously sat- urated by 4 M ammonium sulfate for 1 h. The precipitated protein in this step was removed by centrifugation at 18,000 rpm for 20 min. The supernatant was loaded onto a HiTrap phenyl Sepharose column (GE Healthcare Bio-Science) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.5) supplemented with 2.0 M ammonium sulfate. After washing off unbound proteins, the bound pro- teins were eluted with a linear gradient of 2.0-0.0 M ammonium sulfate in the same buffer. The active fractions were pooled and dialyzed against 20 mM Tris- HCl buffer (pH 7.5) over night. After dialysis, the supernatant was loaded onto a HiTrap Q column (GE Healthcare Bio-Science) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.5), and eluted with a gradient of 0.0-1.0 M sodium chloride in the same buffer. The active fractions were pooled and dialyzed, as described above. The supernatant was loaded onto a Resource Q column (GE Healthcare Bio-Science) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.5), and eluted with a linear gradient of 0.0-0.25 M NaCl in the same buffer. Finally, the enzyme was loaded onto a Superdex 200 pg column (GE Health- care Bio-Science) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.5) containing 0.2 M sodium chloride. The eluted enzyme was pooled and dialyzed against 20 mM Tris-HCl buffer (pH 7.5). All solutions and samples used for purification procedures were filtered through a 0.22-μm or 0.45-μm membrane (Advantec, Tokyo, Japan) to remove dust and any other solids. All the purifi- cation procedures were carried out at 0-4°C. The purity of the protein was con- firmed by SDS-PAGE (Laemmli, 1970).
Identification of the enzyme reaction products
To identify the reaction product of G3DH, the enzyme reactions were carried out by using methylphenazinium methylsulfate (PMS) as an artificial electron acceptor at 37°C under the dark condition. After 6 h of reaction, the re- action mixture was deionized with Diaion SK1B (H+ form, Mitsubishi Chemical, Tokyo, Japan) and Amberlite IRA-411 (Co32- form, Dow Chemical, Midland, MI) ion-exchange resins. The reaction product was purified by CARBOSep COREGEL-87C column (Transgenomic Inc., CA) and was analyzed by NMR. NMR spectra were recorded on a JEOL JNM-ECA 600 FT NMR spectrometer (JEOL, Tokyo, Japan) at 600 MHz for 1H, and 100 MHz for 13C at 30°C. Chemi- cal shifts were referenced to trimethylsilyl propionate (TSP), which is an internal standard. 1H and 13C resonance assignments were made using a combination of two-dimensional NMR homo- and hetero-nuclear correlated spectra (COSY, HMQC, and HMBC). These 2D-NMR spectra were recorded using standard pulse sequences and parameters.
Enzyme characterization
The effect of temperature on enzyme activity was determined using the standard assay conditions but varying temperature from 20 to 70°C. The thermal stability of G3DH was determined by measuring its half-life. The enzyme was incubated at 40 and 45°C, then the residual enzyme activity was measured every 30 min for 3 h. The effect of pH on G3DH activity was determined the standard assay conditions but varying in the pH value from 6.0 to 11.0. The buffer systems were sodium phosphate buffer (pH 6.0-8.0), Tris-HCl buffer (pH 7.5-9.0), and glycine-NaOH buffer (pH 9.0-11.0). For pH stability determination, the purified G3DH was pre-incubated in various buffer systems at the concentration of 20 mM at 4°C for 4 h using the following buffers; citrate buffer (pH 3.0-4.0), acetate buffer (pH 4.0-6.0), sodium phosphate buffer (pH 6.0-8.0) Tris-HCl buffer (pH 7.5-9.0), and glycine-NaOH buffer (pH 9.0-11.0). Then, these enzyme solutions were used for the enzyme activity assay in the standard condition.
To investigate the effect of metal ions on G3DH activity, NiCl2, LiCl2, KCl, MgCl2, CaCl2, FeSO4, CuCl2, MnCl2, ZnCl2, NaCl, CoCl2 and EDTA were used in this experiment. The reaction mixture containing various metal ions at the concentration of 1 mM in 20 mM Tris-HCl buffer (pH 7.5) was incubated at 4°C for 4 h before assay using the standard assay conditions. For EDTA-treated sample, the enzyme solution was dialyzed against 20 mM Tris-HCl buffer (pH 7.5) after incubation. Various monosaccharides, disaccharides and saccharide derivatives at the concentration of 50 mM were used as the substrate in the stan- dard activity assay conditions for the substrate specificity determination.
Finally, the kinetic parameters of G3DH were determined using a Hanes- Woolf plot (Hanes, 1932) in standard reaction condition. The substrates used for kinetic parameters calculation were cellobiose, gentiobiose, lactose, maltose, malto-triose, malto-tetraose, malto-pentaose, malto-hexaose, sucrose, trehalose, and D-glucose. The calculation of kcat used a value of 67,000 as the single sub- unit molecular weight.
D-allose production using G3DH
D-allose production was carried out by a three-step process of enzymatic- dehydrogenation, chemical reduction, and acid-hydrolysis, using cellobiose as the starting material. First, partial purified enzyme containing G3DH was im- mobilized onto IRA-958 ion-exchange resin (Dow Chemical) previously equil- ibrated overnight with 20 mM Tris-HCl buffer (pH 7.5). Then, the immobilized G3DH was added to a reaction mixture containing 50 mM cellobiose and 20 mM PMS before incubation at 30°C under dark condition. The conversion of cellobiose to 3-ketocellobiose was monitored at the indicated times using HPLC. The resultant mixture was evaporated to dryness and re-dissolved in methanol. Then, the sample was reduced by adding 10 mg NaBH4 to the solution and was stirred for 30 min on ice. The reduction was stopped by adding SK1B (H+ form) ion-exchange resin and the resin was removed by filtration. In the final step, the solution was acid-hydrolyzed with 1 M HCl at 100°C for 1 h.
The products from chemical hydrogenation were determined using a Shimadzu HPLC system with a separation column of CARBOSep CORE- GEL-87C. The eluent used was distilled water and its flow rate was 0.6 mL min−1.
RESULTS
Enzyme purification
Rhizobium sp. L35 produced G3DH in the presence of inducing carbo- hydrates such as cellobiose, maltitol, lactose, and lactitol (data not shown). Both ME medium containing 1.0% cellobiose and YE medium containing 1.0% mal- titol were found to be suitable cultivation conditions for the efficient produc- tion of G3DH. The latter was chosen for subsequent experiments because of its cost performance. G3DH was purified approximately 150-fold and to 7.2% yield from crude extract.
Identification of the enzyme reaction product
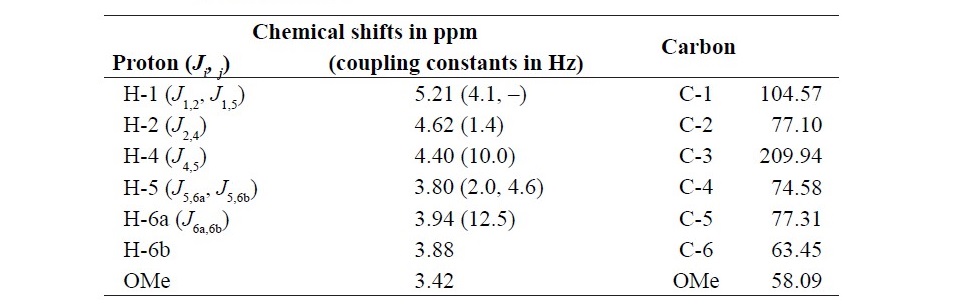
The reaction specificity of the enzyme was confirmed by NMR. Table 1 shows assigned 1H and 13C chemical shifts. A signal from H-3 was absent, and the signal from C-3 was a carbonyl group at 209.94 ppm. Therefore, we deter- mined that G3DH reacts site selectively with the hydroxyl group at the C-3 po- sition of the glycoside residue. From these results, we conclude that the enzyme isolated from Rhizobium sp. L35 belongs to the GMC oxidoreductase family
Table 1. 1H and 13C NMR data for C-3 oxidized methyl α-D-glucopyranoside (methyl-3-keto-α-D-glcp). The chemical shift at the oxidized position is shown in bold.
Enzyme characterization
Purified G3DH showed a band estimated to be 67,000 Da by SDS- PAGE, and its molecular weight was calculated to be 131,000 by size-exclu- sion chromatography, suggesting that it is a dimeric enzyme (data not shown). The optimal temperature for enzyme activity was determined to be 37-40°C, and the half-life of G3DH was approximately 3 h at 40°C and 50 min at 45°C (Figure 2a, b). The optimum pH for enzyme activity was 7.0-7.5, and the enzyme was stable between pH 6.0 and 11.0 (Figure 2c and Figure 2d)
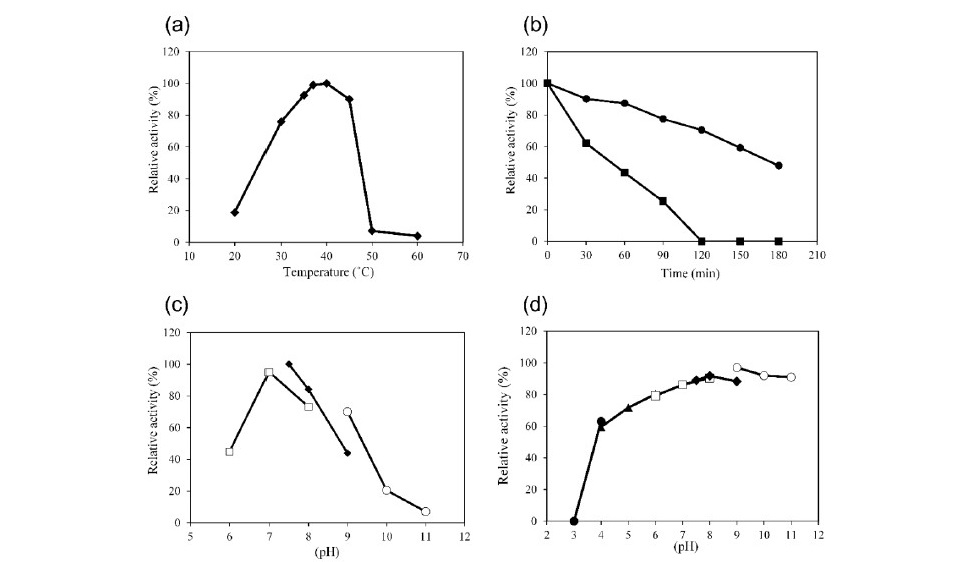
Figure 2. Characteristics of G3DH. Effects of temperature on G3DH activity (a) and stability (b). Effects of pH on G3DH activity (c) and stability (d). The symbols in panels c and d represent citrate buffer (pH 3.0–4.0; ●), acetate buffer (pH 4.0–6.0; ▲), sodium phosphate buffer (pH 6.0–8.0;□), Tris-HCl buffer (pH 7.5–9.0; ♦), and glycine-NaOH buffer (pH 9.0–11.0; ○), respectively.
Most of the investigated metal ions did not affect G3DH activity, how- ever, the activity was slightly enhanced by adding 1 mM FeSO4 to the reaction mixture (15% increase). Activity was inhibited by the addition of CuCl2 and MnCl2 (Figure 3). The purified enzyme had absorption maxima at 279, 391, and 457 nm (data not shown), suggesting that FAD serves as a co-factor for G3DH, as expected for a member of the GMC oxidoreductase family.
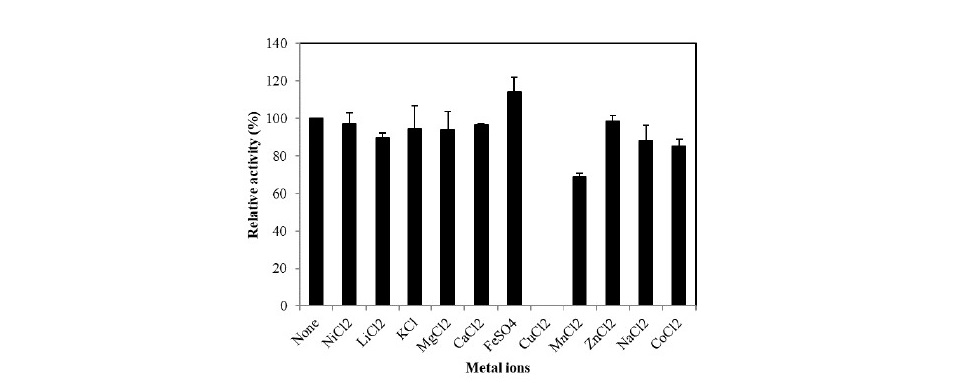
Figure 3. Effects of metal ions on G3DH activity.
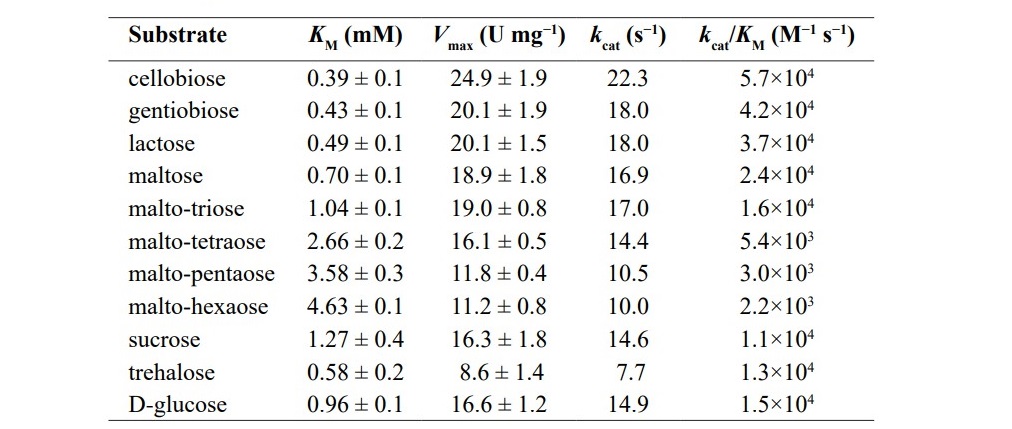
In terms of substrates, the enzyme showed its highest activity toward cellobiose (21.5 U mg-1), followed by salicine [2-(hydroxymethyl)-phenyl β-D-glucose] (19.3 U mg-1), lactose (18.2 U mg-1), maltose (17.8 U mg-1), gen- tiobiose (16.9 U mg-1), maltotriose (16.2 U mg-1), and D-glucose (13.6 U mg-1) as shown in Table 2. This enzyme exhibited low activity towards D-xylobiose (β1,4-D-xylosyl-D-xylose; 1.5 U mg-1) and D-mannobiose (β1,4-D-mannosyl- D-mannose; 1.1 U mg-1). While G3DH showed almost no activity towards D- xylose and D-mannose.
The kinetic parameters of G3DH for various substrates are listed in Table 3. The maximum reaction rate (Vmax), Michaelis-Menten constant (KM), and cata-lytic efficiency (kcat /KM ) for cellobiose were 24.9 U mg-1, 0.39 mM, and 5.7×104 M-1 s-1, respectively, while those for D-glucose were 16.6 U mg-1, 0.96 mM, and 1.5×104 M-1 s-1, respectively. Disaccharides of D-glucose showed 1.3 to 2.4-fold higher affinity than that of D-glucose.
Table 2. Substrate specificity of G3DH from Rhizobium sp. L35.
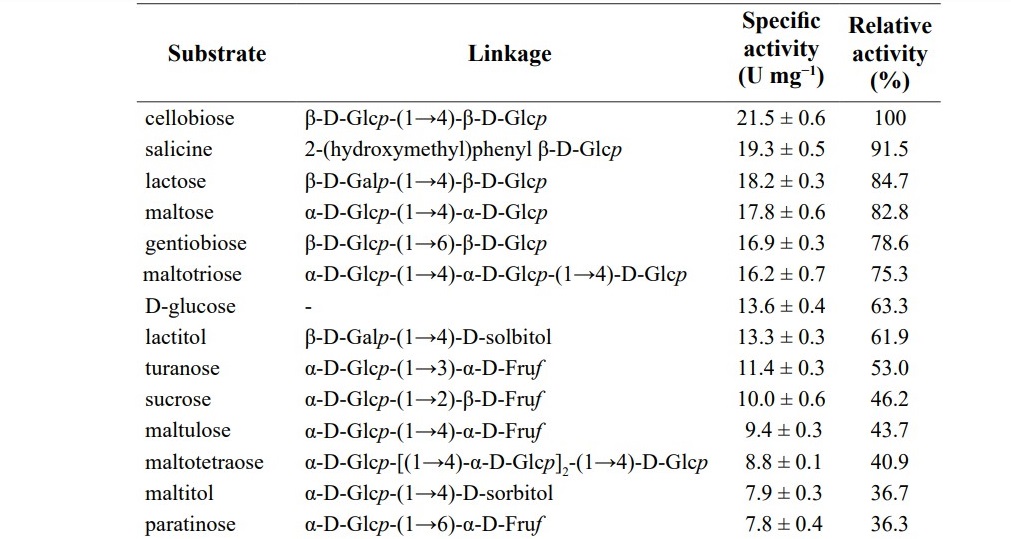

Table 3. Kinetic parameters of G3DH from Rhizobium sp. L35 for various glycosides.
Production of D-allose from cellobiose via 3-ketocellobiose
D-allose could be produced from cellobiose (Figure 4a) by the follow- ing steps. First, the dehydrogenation of cellobiose was done with immobilized G3DH and PMS as an electron acceptor. The reaction was accomplished within 4 h and repeated once. As a reaction product, 3-ketocellobiose (Figure 4b) was achieved before it was reduced by chemical hydrogenation. After the hydrogena- tion, two products were obtained in a ratio of 4:6 estimated by HPLC chromato- gram. The first product (RT 7.9 min) was cellobiose and the other one (RT 8.9 min) was not identical to any known substance. Because of the hydrogenation, the second peak must be a disaccharide consisted of D-glucose and D-allose, which was identified as β-1,4-D-allosyl-D-glucose (Figure 4c). Finally, the ratio of D-glucose (RT 15.6 min) and D-allose (RT 22.3 min) by acid hydrolysis of hydrogenated mixture was estimated to be 3:1 (Figure 4d). Thus, the final yield of D-allose was approximately 30% from cellobiose.
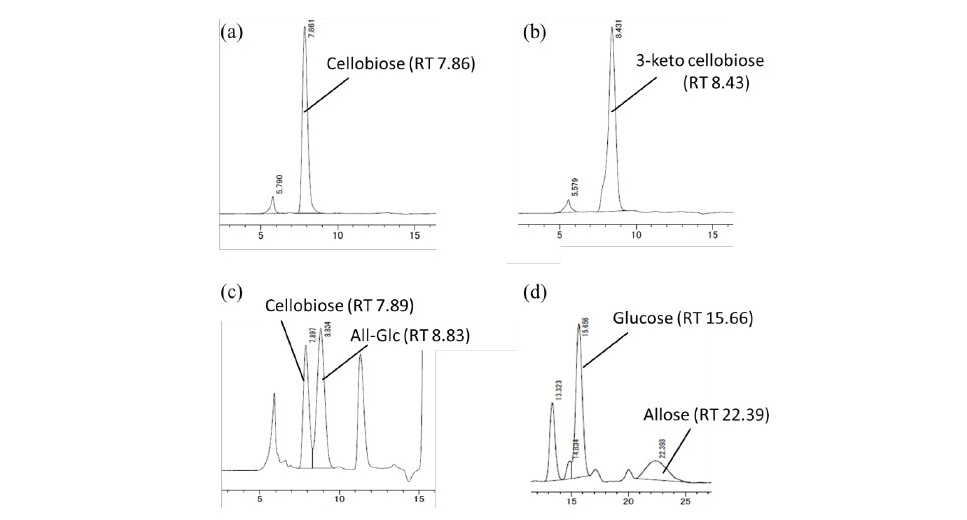
Figure 4. Progress chromatogram of D-allose production. Cellobiose (a) was converted to 3-ketocellobiose (b). Subsequently, 3-ketocellobiose was reduced to cellobiose and allosyl-glucose (All-Glc; c). These two re- sultant products were acid-hydrolyzed to glucose and allose (d). The samples were analyzed by HPLC using a CARBOSep COREGEL-87C column(a–c), anda GL-C611column(d). RTmeansretentiontime(min).
DISCUSSION
In this study, Rhizobium sp. L35 possesses G3DH that can produce 3- ketoglycosides efficiently was isolated from soil. This enzyme was clarified as a member of GMC oxidoreductase family by several experiments. Rhizobium sp. L35 was found to produce G3DH when cellobiose or lactitol was added in the culture medium, therefore, G3DH was identified as an inducible enzyme which is similar to those of A. tumefaciens (Hayano et al., 1967), A. bisporus (Morrison et al., 1999), and Halomonas sp.α-15 (Kojima et al., 1999).
G3DH was purified 150-fold with the total yield of 7.2%. The result of SDS-PAGE and size-exclusion chromatography suggests that G3DH is a di- meric enzyme. Interestingly, most of the G3DHs were reported as a monomeric enzyme such as that of A. tumefaciens (Hayano et al., 1967), Halomonas sp.α-15 (Kojima et al., 1999), A. bisporus (Morrison et al., 1999), or F. saccharophilum (Takeuchi et al., 1986). Only G3DH from F. saccharophilum has been reported as a tetrameric enzyme (Takeuchi et al., 1988). The molecular weight of a single subunit of G3DH from Rhizobium sp. L35 is similar to other G3DHs which is in the range of 55,000-68,000 when estimated by SDS-PAGE (Jin et al., 2008). However, the molecular weights in vitro are different.
The optimum temperature for G3DH activity was 37–40°C, which is 10–20°C higher than other known G3DH (Jin et al., 2008). G3DH tended to show higher activity towards D-glucose and various disaccharides consisting of D-glucose, which is also observed in other G3DHs (Takeuchi et al., 1988 and Zhang et al., 2014). All the hydroxyl groups of D-glucose are in equatorial po- sitions except for the C-1 hydroxyl group, while its epimers, D-mannose, D-al- lose, or D-galactose, has axial hydroxyl groups at C-2, C-3, or C-4. The substrate specificity for these monosaccharides suggests that axial hydroxyls group induce steric hindrance, which decreases enzyme activity at the C-3 (no activity), C-2 (trace activity), or C-4 (2.6 U mg−1) position. As described above, G3DH exhib- ited almost no activity towards D-mannose due to steric hindrance from the C-2 axial hydroxyl group of the sugar. However, β-1,4-D-mannobiose can be recog- nized as a substrate. Similar results were found for D-xylose and β-1,4-D-xylo- biose. A disaccharide substrate is likely to be recognized and bound more easily by the active site compared with a monosaccharide. These two results implied that disaccharides are good matched to the shape of the active site. By using X-ray structural data of G3DH, it may be possible to improve the enzyme’s reac- tivity toward epimers of D-glucose, and make it possible to produce various rare sugars efficiently. Unfortunately, there are no reports on X-ray crystal structural analysis of G3DH, therefore, we are very interested in the structure analysis of this enzyme.
Finally, we succeeded D-allose production by enzymatic reaction. Rhizobium sp. L35 can utilize cellobiose quickly because it produces not only G3DH but also produces various glycoside-degrading enzymes. For this reason, it is difficult to produce sufficient 3-ketocellobiose by the resting cell reaction without by-products (Klekner et al., 1989). The enzyme reaction was carried out using immobilized G3DH. Crude extract was partially purified to remove β-glu- cosidase, a major cellobiose degrading enzyme in Rhizobium sp. L35. D-Allose could be produced approximately 25% yield from cellobiose, which is higher than the conventional method (Granström et al., 2004). This new process of D-allose production can apply for various rare sugars production.
CONCLUSION
Rhizobium sp. L35 with an ability to produce G3DH, an inducible flavine enzyme, was cultivated and the enzyme was then extracted. G3DH was purified in four steps to 150-fold with 7.2% recovery and identified as a homo-dimeric enzyme with a molecular weight of approximately 67,000 Da for a single sub- unit. The optimum condition for G3DH is at 40°C and pH 7.5 in Tris-HCl buffer. The purified G3DH also shown a broad range of substrate specificity. With a new strategy for D-allose production, partial purified G3DH was immobilized before utilized in cellobiose conversion reaction. In the final process, the yield of D-allose was as high as 30% from starting material. Hence, the combination of enzymatic reaction and chemical reaction is an alternative pathway to improve the D-allose productivity.
ACKNOWLEDGEMENTS
This study was supported in part by KAKENHI number 26450097, 22580088 (Grant-in-Aid for Scientific Research (C)).
REFERENCES
Bernaerts, M.J., and De Ley, J. 1963. A biochemical test for crown gall bacteria.
Nature. 197: 406–407. https://doi.org/10.1038/197406b0
Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of micro- gram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 72: 248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Dische, Z., and Borenfreund, E. 1951. A new spectrophotometric method for the detection and determination of keto sugars and trioses. Journal of Biological Chemistry. 192(2):583–7.
Eltz, R.W. 1968. Food antioxidants. U.S. Patent No. 3,372,036.
Giffhorn, F. 2000. Fungal pyranose odidases: occurrence, properties and bio- technical applications in carbohydrate chemistry. Applied Microbiology and Biotechnology. 54: 727–747.
Granström, B.T., Takata, G., Tokuda, M., and Izumori, K. 2004. Izumoring: A novel and complete strategy for bioproduction of rare sugars. Journal of Bioscience and Bioengineering. 97: 89–94. https://doi.org/10.1016/ S1389-1723(04)70173-5
Hanes, C.S. 1932. Studies on plant amylases: The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochemical Journal. 26(5): 1406-1421. https://doi.org/10.1042/bj0261406
Hayano, K., and Fukui, S. 1967. Purification and properties of 3-ketosu- crose-forming enzyme from the cells of Agrobacterium tumefaciens. Journal of Bacteriology. 101(3): 692–697.
Jin, L.Q., and Zheng, Y.G. 2008. Properties of glucoside 3-dehydrogenase and its potential applications. African Journal of Biotechnology. 7(28): 4843–4849.
Kasiganesan, H., Wright, G.L., Chiacchio, M.A., and Gumina, G. 2009. Novel L-adenosine analogs as cardioprotective agents. Bioorganic & Medicinal Chemistry. 17: 5347–5352. https://doi.org/10.1016/j.bmc.2008.12.011
Klenkner, V., Löbl, V., Šimova, E., and Novák, M. 1989. Conversion of disac- chrarides to 3-ketodisaccharides by nongrowing and immobilized cells of Agrobacterium tumefaciens. Folia Microbiologica. 34: 286–293. https:// doi.org/10.1007/BF02814470
Kojima, K., Tsugawa, W., Hamahuji, T., Watazu, Y., and Sode, K. 1999. Effect of growth substrates on production of new soluble glucose 3-dehydro- genase in Halomonas (Deleya) sp. α-15. Applied Biochemistry and Bio- technology. 77–79: 827–834.
Kojima, K., Tsugawa, W., and Sode, K. 2001. Cloning and expression of glu- cose 3-dehydrogenase from Halomonas sp. α-15 in Escherichia coli. Biochemical and Biophysical Research Communications. 282: 21–27. https://doi.org/10.1006/bbrc.2001.4511s
Kujawa, M., Volc, J., Halada, P., Sedmera, P., Divne, C., Sygmund, C., Leitner, C., Peterbauer, C., and Haltrich, D. 2007. Properties of pyra- nose dehydrogenase purified from the litter-degrading fungus Agaricus xanthoderma. FEBS Journal. 274: 879–894. https://doi.org/10.1111/ j.1742-4658.2007.05634.x
Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227(5259): 680–685.
Levin, G.V., Zehner, L.R., Saunders, J.P., and Beadle, J.R. 1995. Sugar substi- tutes: their energy values, bulk characteristics, and potential health bene- fits. American Journal of Clinical Nutrition. 62(5): 11615–11685. https:// doi.org/10.1093/ajcn/62.5.1161S
Maeda, A., Adachi, S., and Matsuo, R. 2001. Improvement of selectivity in 3-ke- tocellobiose production from cellobiose by Agrobacterium tumefaciens. Biochemical Engineering Journal. 8: 217–221. https://doi.org/10.1016/ S1369-703X(01)00108-5
Morrison, S.C., Wood, D.A., and Wood, P.M. 1999. Characterization of a glucose 3-dehydrogenase from the cultivated mushroom (Agaricus bisporus). Applied Microbiology and Biotechnology. 51: 58–64. https:// doi.org/10.1007/s002530051363
Moyroud, E., and Strazewski, P. 1999. L-Ribonucleosides from L-xylose. Tetrahedron. 55: 1277–1284. https://doi.org/10.1016/S0040-4020(98)011 19-3
Sanger, F., Nicklen, S., and Coulson, A.R. 1977. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 74(12): 5463–5467. https:// doi.org/10.1073/pnas.74.12.5463
Takeuchi, M., Ninomiya, K., Kawabata, K., Asano, N., Kameda, Y., and Matsui, K. 1986. Purification and properties of glucoside 3-dehydroge- nase from Flavobacterium saccharophilum. Journal of Biochemistry. 100(4):1049–55.
Takeuchi, M., Asano, N., Kameda, Y., and Matsui, K. 1988. Purification and properties of soluble D-glucoside 3-dehydrogenase Flavobacterium saccharophilum. Agricultural and Biological Chemistry. 52(8): 1905– 1912. https://doi.org/10.1080/00021369.1988.10868964
Takeuchi, M., Asano, N., Kameda, Y., and Matsui, K. 1988. Physiological role of glucoside 3-dehydrogenase and cytochrome c551 in the sugar oxidiz- ing system of Flavobacterium saccharophilum. Journal of Biochemistry. 103: 938–943.
Tsugawa, W., Horiuchi, S., Tanaka, M., Wake, H., and Sode, K. 1996. Purifi- cation of a marine bacterial glucose dehydrogenase from Cytophaga marinoflava and its application for measurement of 1,5-anhydro-D- glucitol. Applied Biochemistry and Biotechnology. 56: 301–310. https:// doi.org/10.1007/BF02786960
Volc, J., Kubátová, E., Wood, D.A., and Daniel, G. 1997. Pyranose 2-dehydro- genase, a novel sugar oxidoreductase from the basidiomycete fungus Agaricus bisporus. Archives of Microbiology. 167: 119–125. https://doi. org/10.1007/s002030050424
Volc, J., Sedmera, P., Halada Přikrylova, V., and Daniel, G. 1998. C-2 and C-3 oxidation of D-Glc, and C-2 oxidation of D-Gal by pyranose dehydro- genase from Agaricus bisporus. Carbohydrate Research. 310: 151–156.
Volc, J., Kubatova, E., Daniel, G., Sedmera, P., and Haltrich, D. 2001. Screening of basidiomycete fungi for the quinone-dependent sugar C-2/C-3 oxidoreductase, pyranose dehydrogenase, and properties of the enzyme from Macrolepiota rhacodes. Archives of Microbiology. 176(3): 178– 86.
Yamaguchi, F., Kamitori, K., Sanada, K., Horii, M., Donga, Y., Sui, L., and Tokuda, M. 2008. Rare sugar D-allose enhances anti-tumor effect of 5-fluorouracil on the human hepatocellular carcinoma cell line HuH-7. Journal of Bioscience and Bioengineering. 106(3): 248–252. https://doi. org/10.1263/jbb.106.248
Zhang, J.F., Zheng, Y.G., Xue, Y.P., and Shen, Y.C. 2006. Purification and char- acterization of the glucoside 3-dehydrogenase produced by a newly isolated Stenotrophomonas maltrophilia CCTCC M 204024. Applied Microbiology and Biotechnology. 71: 638–645. https://doi.org/10.1007/ s00253-005-0201-9
Zhang, J., Chen, W., Ke, W., and Chen, H. 2014. Screening of a glucoside 3-de- hydrogenaseproducing strain, Sphingobacterium faecium, based on a high-throughput screening method and optimization of the culture condi- tions for enzyme production. Applied Biochemistry and Biotechnology. 172: 3448-3460. https://doi.org/10.1007/s12010-0
Akkharapimon Yotsombat1, Keiko Uechi2, Shunsuke Onishi1, Kohei Mino1, Tae Hasegawa1, Kenji Morimoto1, and Goro Takata1*
1Faculty of Agriculture, Kagawa University, 2393 Ikenobe, Miki-cho, Kita-gun, Kagawa 761-0795, Japan
2Faculty of Agriculture, University of the Ryukyus, 1 Senbaru, Nishihara-cho, Nakagami-gun, Okinawa 903-0213, Japan
*Corresponding author. Email: goro@ag.kagawa-u.ac.jp
Total Article Views
Article history:
Received: July 16, 2018;
Revised: October 25, 2018;
Accepted: October 29, 2018