 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Development of a Reverse Transcription-Recombinase Polymerase Amplification Assay for Duplex Detection of Foodborne Viruses in Oysters
Palatip Chutoam, Suthasinee Jinda, Supannee Lethochavalit, Uraiwan Intamaso*Published Date : 2023-01-05
DOI : https://doi.org/10.12982/NLSC.2023.008
Journal Issues : Number 1, January-March 2023
Abstract Viral contamination may occur at any stage of food processing. The study aim was to develop a two-step reverse transcription (RT)-recombinase polymerase amplification (RPA) assay and evaluate for in-field duplex detection of hepatitis A virus (HAV) and norovirus in oysters. The RNA expression plasmids were generated by amplifying a fragment of the VP1 gene of HAV and norovirus through PCR and cloning it into an expression vector. The RNAs were transcribed in vitro from the plasmids and further used for reverse transcription. The resulting single-stranded cDNAs were used as the purified or spiking templates to determine the sensitivities of simplex and duplex RT-RPA compared with RT-PCR, and RT-qPCR assays. The reproducibility and application of duplex RT-RPA in the field were also evaluated. Our results showed that simplex RT-RPA was at least 100 times more sensitive than RT-PCR and RT-qPCR and even more than duplex detection using purified targets. Unlike RT-PCR, the RT-RPA reaction was unaffected by inhibitors found in food, allowing simple sample preparation methods for detection within a fraction of the time. The duplex assay detected HAV, norovirus, or both in 12/30 (40%) oyster samples tested. Duplex RT-RPA proved to be a rapid, accurate, and reproducible method in a field test for detecting HAV and norovirus. Thus, duplex RT-RPA should be suitable for use in minimally equipped laboratories and field settings. If in-field RT-RPA services are provided to oyster farmers, the technique can minimize the risk of infection to consumers, thereby improving food safety.
Keywords: Food safety, Hepatitis A virus, Direct extraction, Norovirus, Nucleic acid amplification
Funding: This research project was financially supported by Agricultural Research Development Agency (Public Organization) or "ARDA," grant number 6305031040.
Citation: Chutoam, P., Jinda, S., Lethochavalit, S., and Intamaso, U. 2023. Development of a reverse transcription-recombinase polymerase amplification assay for duplex detection of foodborne viruses in oysters. Nat. Life Sci. Commun. 22(1): e2023008.
INTRODUCTION
Fresh and minimally processed foods are a known potential vehicle for foodborne viruses. When cultured in polluted water, oysters can be a significant source of foodborne viruses because they ingest particles from the water (Carter, 2005; Pintó et al., 2009). In addition to during primary production, viral contamination may occur at any of the food processing stages of processing, storage, distribution, and final preparation. The World Health Organization has estimated that 600 million individuals develop foodborne illnesses after eating contaminated food, and 420,000 die every year (World Health Organization, 2022a). Among foodborne viruses, hepatitis A virus (HAV) and norovirus are significant causes of foodborne outbreaks globally. Norovirus is the primary cause of acute gastroenteritis in developing countries, leading to 50,000 child deaths annually (Centers for Disease Control and Prevention (CDC), 2021). On the other hand, HAV increases the risk of acute hepatitis and even causes a greater mortality rate in persons >50 years old, ranging from 1%–5.4% (Lemon et al., 2017), with an estimated 7,134 deaths in 2016 (World Health Organization, 2022b). Monitoring foods for viral contamination at control points along the food supply chain helps prevent food contamination and minimize the risk of foodborne illnesses. Therefore, sensitive and specific methods are required to regularly monitor food contaminations from cultural practices and consumer purchasing sources.
Several factors should be considered aside from sensitivity and specificity when selecting a suitable food-contamination detection method. Although PCR-based assays are highly sensitive, they require specialized technical knowledge and costly equipment (Piepenburg et al., 2006). Furthermore, PCR-based approaches are susceptible to PCR inhibitors present in the food matrix. Additionally, extraction of highly purified nucleic acids from food samples is time-consuming and impracticable in the field. On the other hand, loop-mediated isothermal amplification (LAMP) may detect nucleic acids on-site but requires more effort to design primers (Yoda et al., 2007). Several primers must be designed to bind to six places at a particular distance in the target sequence for conducting the test. The primer design is more difficult for highly variable genome sequences of viruses. The recommended temperature for amplification is 65°C and the reaction temperature typically ranges from 30 to 60 min. In addition, turbidity or color intensity to visualize the results may not be appropriate for multiplex detection.
Another isothermal nucleic acid amplification approach is recombinase polymerase amplification (RPA), which can detect nucleic acids in 15–20 min at a constant temperature of 37°C–42°C (Li et al., 2018) with a lower reaction temperature and only a pair of primer oligonucleotides (Lobato and O’Sullivan, 2018). A reduced need of complicated designed primers and time makes RPA more attractive than LAMP. Unlike PCR, the reactions are unaffected by inhibitors found in food, enabling immediate detection in a very short time (Rajiuddin et al., 2020). Moreover, the sensitivity and specificity of RPA have been shown to be comparable to those of conventional PCR assays (Daher et al., 2016). RT-RPA has been successfully used in the diagnosis of bovine coronavirus, bovine viral diarrhea virus (Aebischer et al., 2014), human norovirus (Han et al., 2020; Moore and Jaykus, 2017), and murine norovirus (Ma et al., 2018). In nature, more than one pathogen might coexist in a single food sample at low concentrations. Hence, it is necessary to identify multiple foodborne pathogens with a speedy, highly sensitive, and specific detection approach. Several investigators have developed multiplex RPA assays to detect foodborne pathogens, such as bacteria in food (Ma et al., 2020; Tran et al., 2022), intestinal protozoa (Crannell et al., 2016), and viruses in clinical samples (Li et al., 2021). So far, only a few reports on multiplex detection of foodborne viruses have been published.
The study aim was to develop a two-step reverse transcription (RT)-RPA for duplex detection of HAV and norovirus and evaluate its performance in field applications by using minimally processed in oyster samples. If in-field RT-RPA services are provided along the food chain, the technique can minimize the risk of infection to consumers, thereby improving food safety.
MATERIALS AND METHODS
Samples
We bought fresh oysters with shells randomly from 2-3 shops selling oysters at Ang sila, Chonburi. After scrubbing their outer surface with a stiff brush and rinsing with water to remove any mud and sand, digestive glands were dissected, homogenized, and pooled into a 2-g sample from each oyster for viral analysis.
Primer designs
On the recommendation of TwistAmp (TwistDX, UK), we designed five forward and reverse RT-RPA primers specific to the conserved region of the coat protein gene, VP1. The Oligonucleotide Properties Calculator (Kibbe, 2007) was used to check the qualification of all HAV and norovirus primers, each of which was 35-bp long. A perfect nucleotide match (100%) was set to ensure the RT-RPA primers’ capability to detect all HAV and norovirus strains known to date. All primers had a guanine-cytosine content between 29% and 57%. All primers were synthesized (Macrogen, Seoul, South Korea) and screened for amplification efficiency. We selected 232- and 205-bp amplified products from norovirus and HAV, respectively, for further use in duplex detection because of their clear separation of the DNA fragments by agarose gel electrophoresis. The chosen primers were also tested for their specificity against non-target enteric viruses, including hepatitis E virus, poliovirus, rotavirus, enterovirus EV71, and coxsackievirus B5.
Construction of RNA expression plasmids
RNA expression plasmids, pET-HAV, and pET-norovirus were created by amplifying a fragment of the VP1 gene of HAV (Accession No. EF207320), and norovirus DNA fragments (Accession No. MG78678) through PCR (Figure 1). Amplified fragments of 422 bp of HAV and 352 bp of norovirus were cloned at 5’XbaI and 3’BlpI restriction sites into pET-28b(+) (SnapGene, San Diego, California, USA) (Fang et al., 2018). Transformants growing on luria agar plates (LA) 50 µg/mL kanamycin (KAN) were selected. All transformants were screened for insert identity by double-enzyme digestion, PCR, and DNA sequencing before expression.
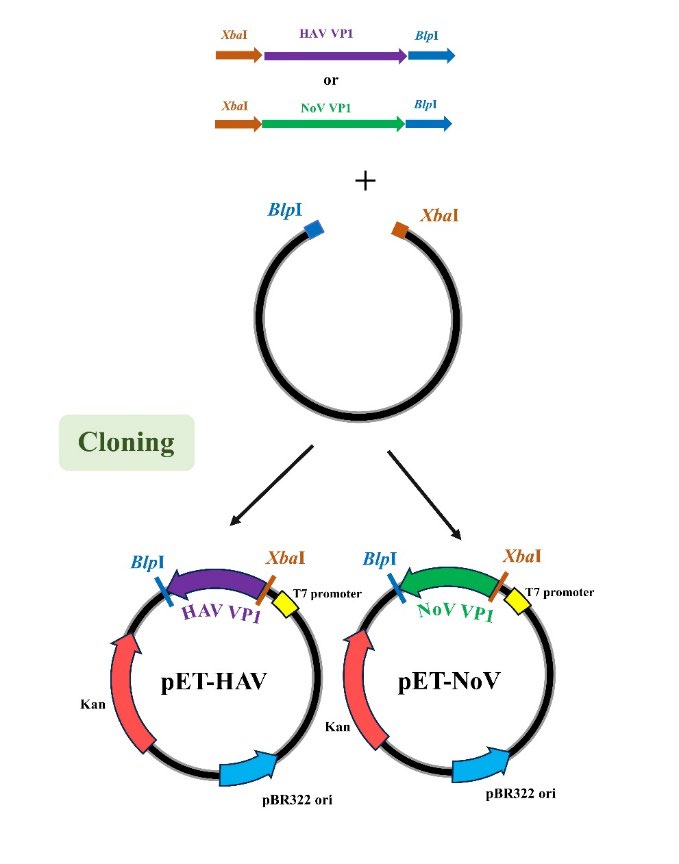
Figure 1. Schematic flow diagram of methods used to construct pET-HAV and pET-norovirus.
Preparation of RNA transcribed in vitro
RNAs were transcribed from pET-norovirus or pET-HAV by subjecting them to a HiScribe T7 Quick High Yield RNA Synthesis kit (New England Biolabs, Hitchin, U.K.) according to the manufacturer’s instructions. The reaction system was 20 µL in total: 2 µL of T7 RNA Polymerase Mix, 10 µL of NTP buffer, and 1 µg of plasmids in 8 µL of dH2O. The reactants were incubated at 37°C for 4 h, followed by DNA elimination with DNase I setting at 37°C for 15 min. The transcribed RNA was purified and quantified. The integrity of RNA products was analyzed by denaturing RNA electrophoresis.
Preparation of denaturing gel electrophoresis for RNA analysis
Eight microliters of transcribed products was mixed thoroughly with 2 µL of 10X MOPS Buffer (200 mM MOPS, 50 mM sodium acetate, 10 mM EDTA∙2H2O, 10 mM EGTA), and 10 µL formamide in a total of 20 µL. The mixtures were heated at 70°C for 10 min and suddenly chilled on ice for ≥1 min before loading. The RNA mixtures were added with 2 µL formaldehyde loading buffer (1 mM EDTA, pH 8.0, 0.4% bromophenol blue, xylene cyanol, and 50% glycerol). The total mixtures were then loaded in 1% agarose gel containing 1X MOPS Buffer and 37% (v/v) formaldehyde and then electrophoresed at 50 V for 60 min. The RNA bands were stained with 1 µg/mL ethidium bromide for 5 min and destained for 40 min.
Reverse transcription (RT)
The first strand cDNAs were reverse transcribed using a Random Hexamer RevertAid First Strand cDNA Synthesis kit (Thermo Scientific, Waltham, Massachusetts, USA) following the manufacturer’s instructions. Briefly, the total RNAs (0.1 ng–5 µg) was mixed with 1 µL of oligo dT Random Hexamer primer, 4 µL of 5X Reaction Buffer, 1 µL of Ribolock RNase inhibitor (20 U/µL), 2 µL of 10 mM dNTPs mix, and 1 µL of RevertAid M-MuLV RT (200 U/µL) in a total volume of 20 µL. The mixture was incubated at 25°C for 5 min, followed by 42°C, 60 min. The reaction was then stopped after sitting at 70°C for 5 min. The resulting single-stranded cDNAs were used as the templates for the downstream RPA, PCR, and qPCR assays. The copy number was calculated according to the following formula:
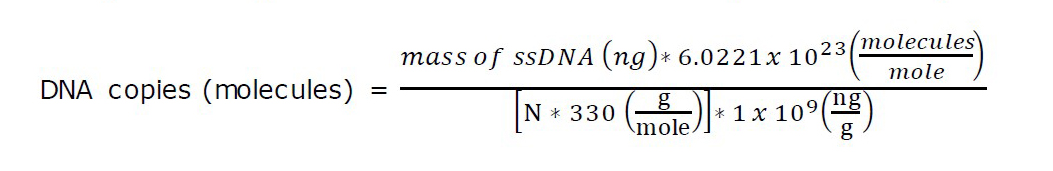
Recombinase polymerase amplification (RPA)
Then, 1 µL of cDNA templates was added to the simplex RPA reaction TwistAmpBasic Kit (TwistAmp Ltd., Cambridge, UK) containing 0.42 µM forward and reverse primers, 29.5 µL TwistAmp rehydrate buffer in a 47.5-µL reaction volume (TwistAmp Ltd., Cambridge, UK.). The reaction was initiated by adding 2.5 µL of 14 mM Mg (Ac)2 to the final reaction volume of 50 µL per sample. In duplex RPA reactions, both sets of 0.7µM HAV and norovirus primers were added to a single reaction containing 50 ng of each template and 18 mM Mg(Ac)2. Those reactions were incubated in a water bath or heat block at 37°C for 20 min. The RPA end products were purified using a GenepHlowTM Gel/PCR Kit (Geneaid, New Taipei City, Taiwan). Then, the purified amplification products were analyzed on agarose gel electrophoresis using 1% (w/v) agarose gels containing 1X SYBRÒ (Invitrogen by Thermo Fisher Scientific, USA) in 1X Tris-Boric acid-EDTA (TBE) buffer.
Conventional PCR
Simplex PCR was performed in 50-µL reactions containing 1 µL of HAV or norovirus cDNAs with 0.2 µM of each primer, 200 µM dNTP mix, 2 mM MgCl2, and 1.25 U Phusion high-fidelity DNA polymerase (Thermo Scientific, USA). The reaction condition was conducted for HAV, 94°C for 5 min for the initial denaturation, followed by 35 cycles of 94°C for 30 s, 70°C for 30 s, 72°C for 1 min and one final extension of 72°C for 5 min. The reaction condition was carried out for norovirus at 94°C for a 5-min initial denaturation, followed by 35 cycles of 94°C for 30 s, 72°C for 1 min, and one final extension of 72°C for 5 min on a thermocycler (T100 Thermal Cycler; Bio-Rad, Hercules, California, USA).
The conditions for duplex PCR were screened by varying the annealing temperature, primer, and template ratios. The optimal duplex PCR was established in 50-µL reactions containing RNAs (50 ng of norovirus and 100 ng of HAV), primers (0.1 µM norovirus primers and 0.2 µM HAV primers), 200 µM dNTP mix, 2 mM MgCl2, and 1.25 U of Phusion high-fidelity DNA polymerase (Thermo Scientific, USA). The following PCR conditions were used: 94°C for 5 min for the initial denaturation, followed by 35 cycles of 94°C for 30 s, 67°C for 30 s, 72°C for 2 min and one final extension of 72°C for 5 min. Amplification products were analyzed by agarose gel electrophoresis using 1% (w/v) agarose gels containing 1X SYBRÒ (Invitrogen by Thermo Fisher Scientific, USA) in 1X TBE buffer.
Quantitative PCR (qPCR)
The Maxima SYBR Green qPCR master mix kit was used for real-time PCR by Applied Biosystems QuantStudio5 (Thermo Scientific, USA). The optimized reaction contained 12.5 µL of 2X Maxima SYBR Green qPCR Master Mix, 0.05 µL ROX solution, 1.5 µL UDG (1 unit/µL), 1.5 µL UDG buffer, 1 µL of cDNA template, 0.75 µL of 10 µM HAV forward and reverse primers or 2.25 µL of 10 µM norovirus forward and reverse primers, and 4.95 µL nuclease-free dH2O in 25 µL total reaction volume. The reaction steps were set as follows: UDG pretreatment at 50°C for 2 min, initial denaturation at 98°C for 3 min, followed by amplification over 35 cycles at a denaturation temperature of 98°C for 10 sec and an annealing temperature of 70°C (HAV) or 72°C (norovirus) for 30 sec. Ten-fold serially diluted plasmids were used to generate the standards. All standards and cDNA samples were run in triplicate, and the whole assay was performed twice. The Ct value, which represents the number of PCR cycles in which the reporter dye fluorescence was detectable above the fluorescence threshold (Ct = 35), was computed automatically for each amplification cycle. The qPCR results were considered positive for Ct > 35.
Sensitivity tests
Serial 10-fold diluted templates were made using the cDNA synthesized from transcribed RNA. One microliter of each diluted template was in RPA and conventional PCR, and their amplified products were analyzed by agarose gel electrophoresis. Band intensities of amplified products on agarose gel were measured in the Image J program (developed by the National Institutes of Health and Laboratory for Optical and Computational instrumentation). The standard curves were generated using the Y-axis for band intensities and the X-axis for copies. Each dilution was tested in duplicate, and the whole assay was performed twice.
Application of RT-RPA to virally contaminated oysters
Initially, all oysters were verified to be free of HAV and norovirus by RT-RPA. In spiking experiments, 2 g of oysters were homogenized with 5 µL of 10-fold diluted of HAV-RNAs or norovirus-RNAs for simplex reactions, and both HAV-RNAs and norovirus-RNAs for duplex detection at 37°C for 1 h. Each spiked sample was then added with 2 µL of 100 µg/mL proteinase K (Biotechrabbit, Hennigsdorf Germany). After two consecutive shaking incubations at 37°C for 1 h and 60°C for 15 min, the crude extracts were cleared by centrifugation at 14,000g for 5 min and filtered by filter papers. RNAs in the extracts were used to amplify cDNAs by RT-RPA and RT-PCR.
Our extraction method was compared with another detailed extraction method that obtains more purified templates (Studier, 2005). All extraction steps were the same for viral contamination testing in oysters but omitted the spiking step. The duplex RT-RPA assays detected all samples. Purified RNAs yielding positive amplification determined by RT-RPA were used as the positive control, whereas the negative control was the oyster extract free of HAV and norovirus. The whole assay was performed twice.
DNA verification by sequencing
To test viral contaminations in oysters, amplified RT-RPA proucts were purified from the agarose gel (GeneHlow Gel/PCR kit; Geneaid, New Taipei City, Taiwan) and the identities confirmed by DNA sequencing (Macrogen, Seoul, South Korea).
Reproducibility of the tests. HAV-RNAs was added to every 2 g of oyster and extracted for RT-RPA reactions. Intra-assay precision was assessed using five replicates per assay on the same day, and inter-assay precision was assessed by performing five assays on different days. The band intensity of RPA products on agarose gel was measured by the ImageJ program and estimated for molecule numbers referenced to the standard curve. Inter- and intra-assay precision were assessed to determine for statistical significance (P < 0.05).
Statistical analysis
The reproducibility of results was evaluated statistically using one-way analysis of variance, and the level of statistical significance was set to P < 0.05. Standard curves were generated and linear regression was used to test for equivalence of band intensities versus quantitation (copies) of RT-RPA versus RT-PCR. Data with a correlation co-efficiency of r2 ≥ 0.9 was considered to be acceptable for analysis.
RESULTS
Construction of plasmids. Expression plasmids, pPET-norovirus, and pPET-HAV were constructed and their identities verified by double digestion, PCR, and DNA sequencing. RNAs were synthesized by in vitro transcription to test their expression ability. All plasmids were transcribed to RNAs, as visualized on 1% denaturing agarose gel electrophoresis (data not shown).
RPA primer screening for RPA sensitivity and specificity. Five potential primer pairings for HAV and norovirus VP1 genes were screened for strong amplification and distinct sizes of products visible on agarose gel electrophoresis (Figure 2 and 3, resepctively). Finally, one primer set was selected for each target region (Table 1). These primers were also used to test their target specificity. Only RT-RPA products were amplified from HAV and norovirus. In contrast, no amplification was observed with other foodborne viruses (Figure 4), which showed no cross-reactivity between the viral pathogens tested.
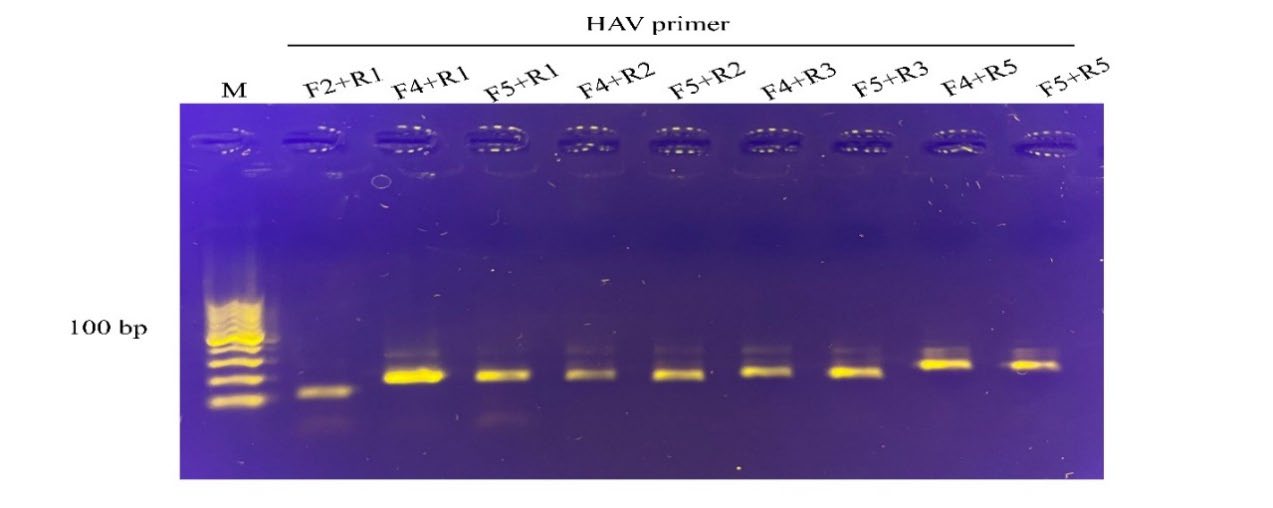
Figure 2. Screening primer pairs for amplification efficiency of the HAV target by RPA assay. The primer pair, F4 and R1, yielding 205 bp amplified product, were selected and further used in the assay. M, DNA marker (100 bp from the bottom to top).
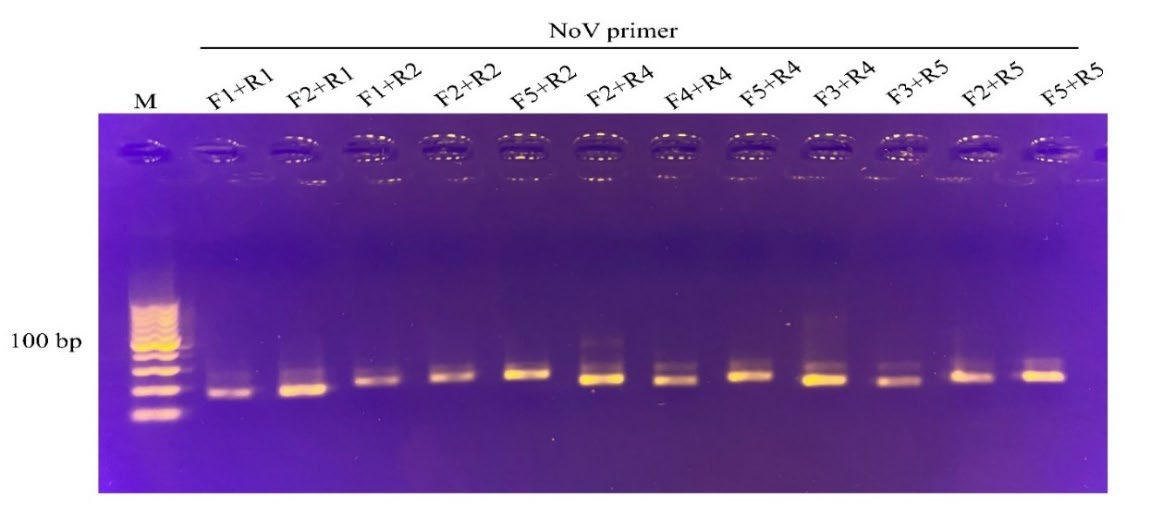
Figure 3. Screening primer pairs for amplification efficiency of the norovirus target by RPA assay. The primer pair, F2 and R2, yielding 232 bp amplified product, were selected and further used in the assay. M, DNA marker (100 bp from the bottom to top).
Table 1. Primer sets used in this study.
|
Oligo name Oligo sequence (5' to 3') Tm (°C) |
||
|
HAV_F4 |
TCTACTGAGCAGAATGTTCCTGATCCCCAAGTCGG |
76.9 |
|
HAV_R1 |
CTGATGTATGTCTAAACTCTCCAGGTTTCAATTCA |
70.9 |
|
NOROVIRUS_F2 |
GCACCTGTAGCGGGCCAACAAAATGTAATTGACCC |
76.9 |
|
NOROVIRUS_R2 |
CTGTGAACGCGTTCCTCACTAGAATTACCTGCACT |
74.9 |
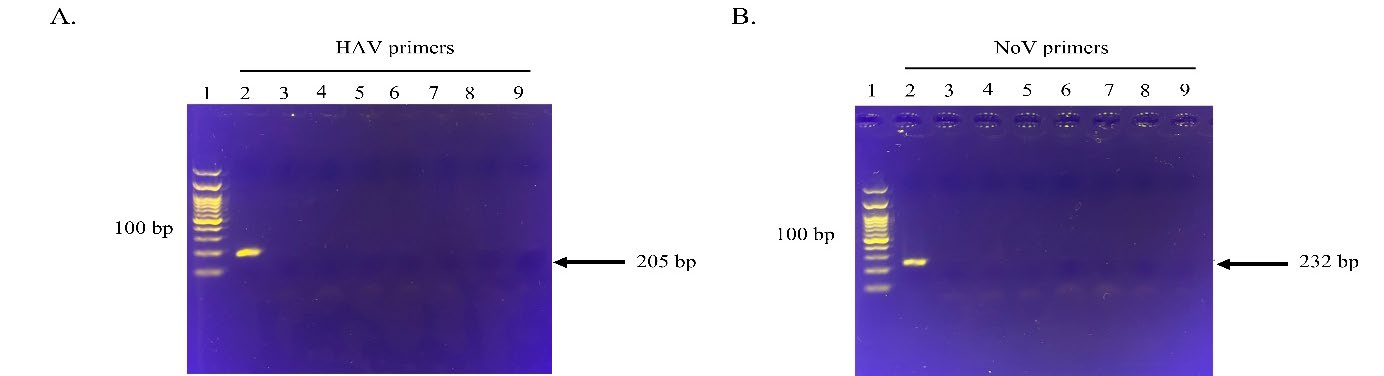
Figure 4. Specificity of the HAV and norovirus primer pairs to their targets. HAV F4 and R1 primers binding to HAV (A) and norovirus F2 and R2 primers binding to norovirus (B). Lane 1, DNA marker (100 bp from the bottom to top); lane 2, HAV and norovirus, respectively; lane3, norovirus and HAV, respectively; lane 4, rotavirus; lane 5, EV71, enterovirus EV71; lane 6, coxsackievirus B5; lane 7, hepatitis E virus; lane 8, poliovirus; lane 9: the negative control without the template. The arrows on the right indicate the expected RPA amplification products of HAV and norovirus, respectively. The images represent results from two independent experiments.
Comparison of simplex RT-RPA, RT-PCR, and RT-qPCR assays. The specified cDNA templates were amplified by RPA and compared with those of the conventional PCR using the same RPA primers. RT-RPA could detect HAV RNAs with 4.54 x 104 copies with visible amplification from the non-diluted sample to a dilution of 1 x 10-8 (Figure 5). However, RT-PCR detected the HAV templates in the range of 107 copies/reaction (Table 2). The findings showed that RT-RPA was at least 1,000 times more sensitive than the conventional RT-PCR assay. The R2 values obtained over 0.98, demonstrating that RT-RPA and RT-PCR were equivalent (Figure 6).
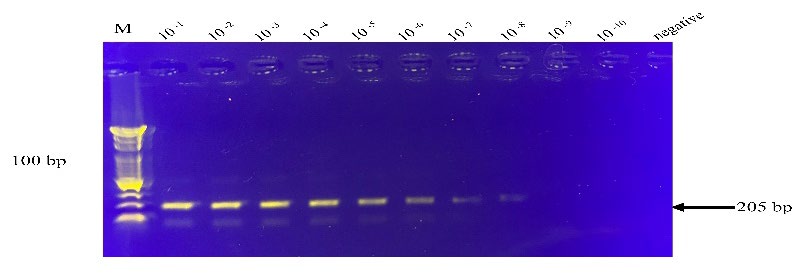
Figure 5. The simplex RT-RPA detection limits for HAV RNA transcripts. The dilutions are indicated on top of individual lanes and the negative control without the template. The images shown are representatives of two independent experiments. The arrow on the right of each figure indicates the expected RT-RPA amplification products of HAV. M, DNA marker (100 bp from the bottom to top).
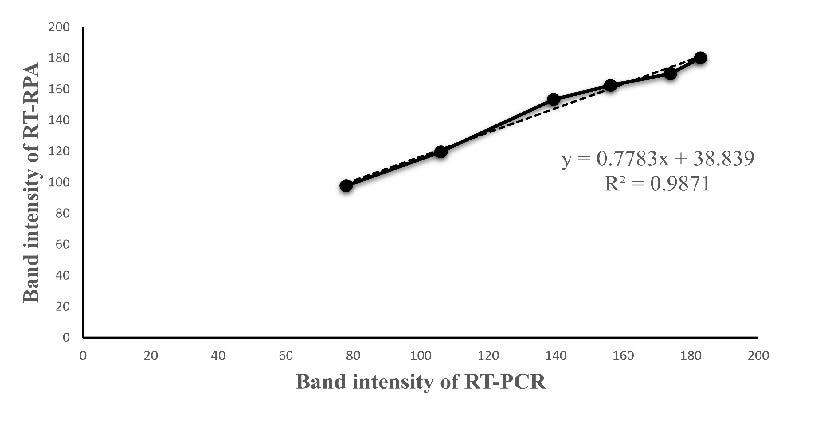
Figure 6. The equivalence of RT-RPA and RT-PCR using ten-fold serial dilutions of HAV RNAs. Band intensities of RT-RPA and RT-PCR amplified products on agarose gel were measured by program J (developed by the National Institutes of Health and Laboratory for Optical and Computational instrumentation). Linear Regress was used to test for the equivalence with the R2 values over 0.98.
To determine the sensitivity of qPCR, 10-fold serially diluted recombinant plasmids were amplified to generate the amplification plot. The standard curve was constructed by plotting the cycle threshold (Ct) value on the Y-axis and the known log-dilution of DNA in 10-fold dilutions on the X-axis. (Figure 7). Our results showed that the primers could detect (Ct values < 35) HAV cDNA as low as 8.90 × 106 copies/reaction and norovirus at 1.26 × 108 copies/reaction. These results indicated that RT-RPA was 100 times more sensitive than RT-qPCR (Table 2).
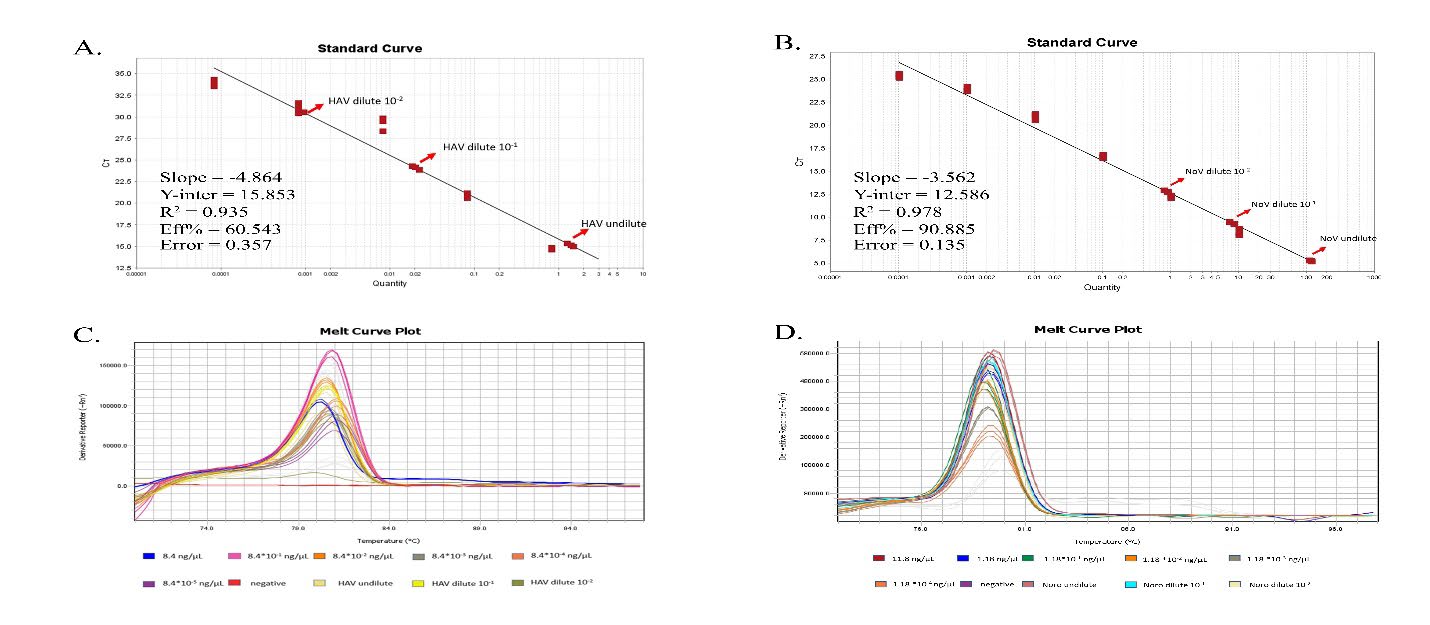
Figure 7. The sensitivity of qPCR. The standard curve generated from the cycle threshold of each of ten-fold serially diluted pET-HAV (A) and pET-norovirus (B). The melt curve plots derived from figure A and B illustrate in C and D, respectively.
Table 2. Comparison of RT-RPA, RT-PCR and RT-qPCR assays.
|
Reaction
|
Methods
|
Targets
|
Purification |
Spiking contamination |
||||||
|
Dilution |
LODa (molecules) |
Dilution |
LODa (molecules) |
|||||||
|
Simplex |
RT-RPA |
HAV |
10-8 |
4.54 × 104 |
10-2 |
1.89x109 (Ma et al., 2018) |
||||
|
RT-PCR |
HAV |
10-6 |
4.54×107 |
10-1 |
1.89x1012 (Ma et al., 2018) |
|||||
|
|
|
|
|
|||||||
|
qRT-PCR |
HAV |
10-1 |
8.90×106 |
|
NDb |
|||||
|
norovirus |
10-2 |
2.36×107 |
|
|
||||||
|
Duplex |
RT-RPA |
HAV |
10-5 |
4.45 × 106 |
|
4.45 × 1012 |
||||
|
norovirus
|
10-5
|
3.93 × 106
|
|
9.83 × 1011
|
||||||
|
RT-PCR |
HAV |
|
8.90 × 1011 |
|
NDb |
|||||
|
norovirus |
|
1.97 × 1011 |
|
|
||||||
|
Note: a LOD, The limit of detection bND, Not determined
|
|
|
|
|
||||||
Comparison of the duplex RT-RPA dectection limits for HAV- and norovirus-RNAs
To test the sensitivity of the duplex RT-RPA, 10-fold serial dilutions of purified RNAs were prepared. At the highest dilution of 1 × 10−5, the cDNA bands were still observed (Figure 8). As a result, the detection limits for the simultaneous detection of HAV- and norovirus-RNAs were as low as 4.45 × 106 and 3.93 × 106 copies/reaction, respectively (Table 2).
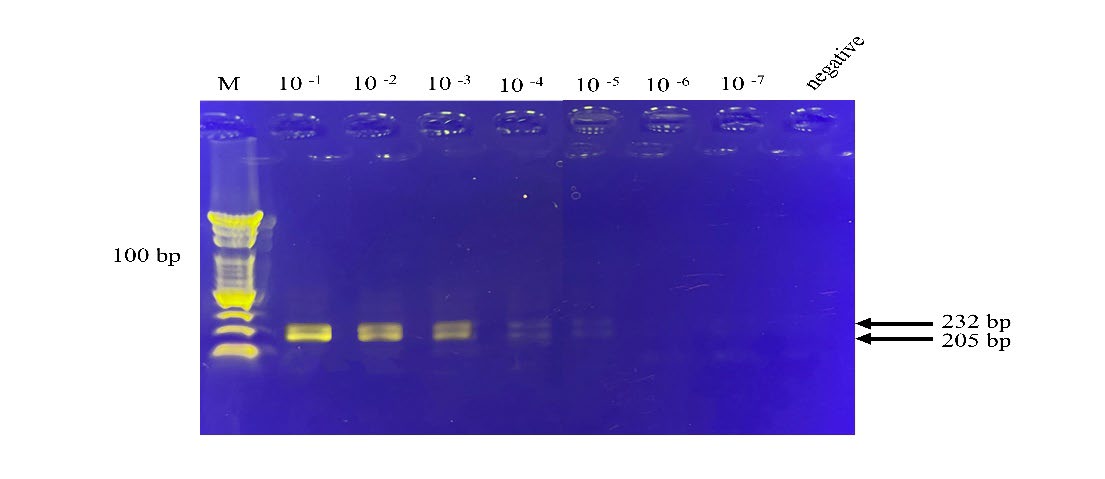
Figure 8. The duplex RT-RPA detection limits for HAV-and norovirus-RNAs. RT-RPA amplified ten-fold serial dilutions of HAV-and norovirus-RNAs. The dilutions are indicated on top of the lane and the negative control without the template. The arrows on the right show the expected RT-RPA amplification products of HAV and norovirus of 205 and 232 bp, respectively. The images represent results from two independent experiments. M, DNA marker (100 bp from the bottom to top).
Quantitation of RPA products. The ImageJ program showed that the band intensities of the cDNAs intensified on agarose gel as the amounts of RNAs were increased and the molecules detected were shown to be equivalent (data not shown), with good correlation between band intensities and starting copies (R2> 0.95) (Figure 9).
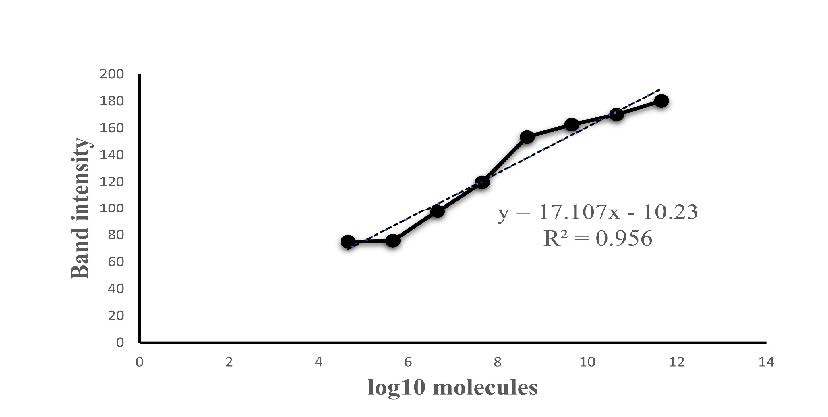
Figure 9. The equivalence of band intensities of RT-RPA amplified products and the number of molecules. RT-RPA amplified ten-fold serial dilutions of purified HAV RNA and their band intensities on agarose gel were measured by program J (developed by the National Institutes of Health and Laboratory for Optical and Computational instrumentation). Linear Regress was used to test for the equivalence with R2 > 0.95.
Reproducibility of the RT-RPA assay
The reproducibly of RT-RPA was also determined by performing the reaction with five samples on each five consecutive days. No statistically significant (P < 0.05) differences were observed between the intra- and inter-reproducibilities (P = 0.45 and P = 0.14, respectively), demonstrating that the RT-RPA assay was highly reproducible.
Spiking experiments
The RT-RPA limit of detection was also tested in spiked food samples. The sensitivity of simplex RT-RPA could not be compared with that of RT-PCR utilizing the direct extraction method because RT-PCR produced amplicons only from purified genetic materials. However, RT-RPA was 1,000 times more sensitive than RT-PCR (1.89 × 109 vs. 1.89 × 1012 copies) when using the alternative extraction method to acquire more refined genetic materials (Table 2).
Field tests
Duplex RT-RPA was evaluated to detect HAV and norovirus contaminations using oyster samples. Their identities were confirmed by DNA sequencing. The results showed that 12/30 (40%) of the oysters were contaminated. HAV was found in 2 (6.67%) of 30 samples, norovirus in 1 (3.33%) of 30 samples, and both HAV and norovirus in 9 (30%) of the oyster samples. Further sequencing results showed that those isolated VP1 gene fragments all belonged to genotype 1 and shared 98%–99% of the homology with the reference HAV LP014 strain (Figure 10) and 89%–92% with the norovirus GII strain (Figure 11), which were the predominant circulating strains in Thailand (Barameechai et al., 2008; Chuchaona et al., 2019).
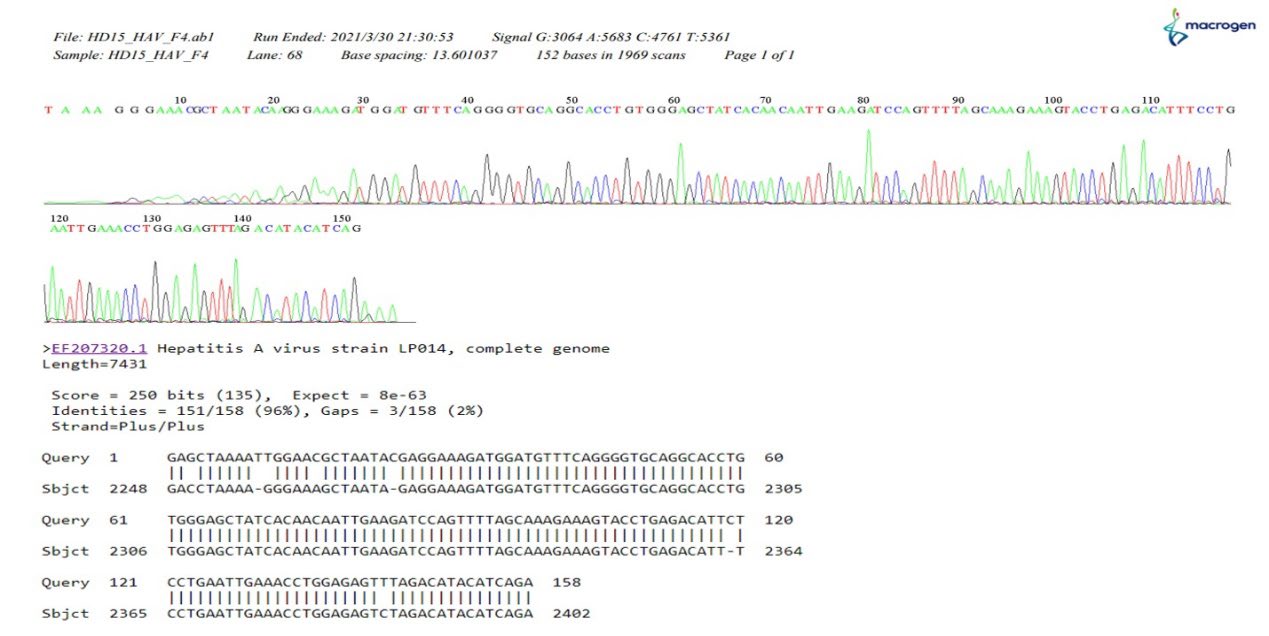
Figure 10. The example of the dendrograms for HAV sequence detected from oyster samples and the matching result with LP014 HAV reference strain.
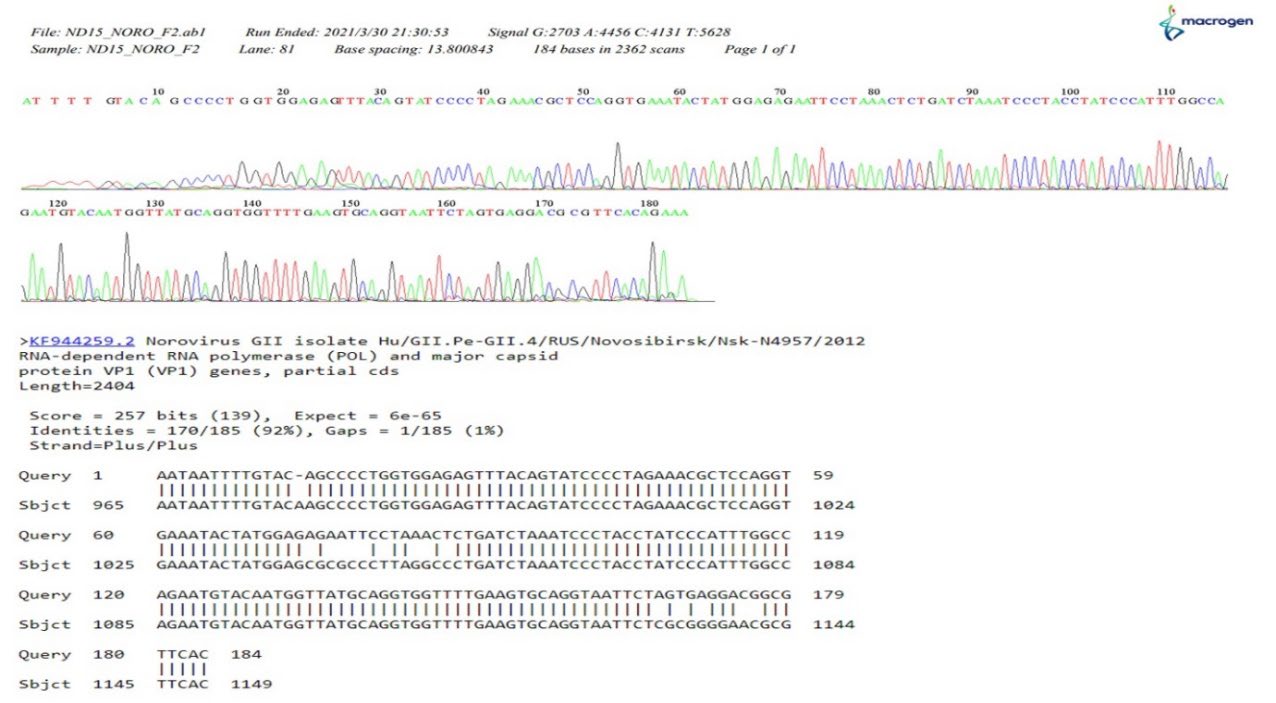
Figure 11. The example of the dendrograms for the norovirus sequence detected from oyster samples and the matching result with GII norovirus reference strain.
DISCUSSION
It is well-known that norovirus and HAV are major enteric viruses linked to foodborne outbreaks that pose food safety risks. In nature, multiple pathogens often coexist in actual food samples. The lack of a multiplex, sensitive, and field-based tool is the main factor that limits routine food safety testing. RT-PCR and RT-qPCR are widely used sensitive techniques for multiplex detection of foodborne pathogens in food and shellfish (Lee et al., 2018). However, their uses in a field setting are restricted because of the need for expensive reagents and equipment (a thermal cycler or a real-time PCR machine), long processing times, and skilled, trained personnel. RT-LAMP is one of the favorite on-site methods because the reaction amplifies the target gene at isothermal conditions. RT-LAMP assays often require six primers, resulting in longer amplicons and making primer design more complicated, especially when identifying RNA viruses with significant genomic diversity (Yoda et al., 2007). We chose the RT-RPA assay over RT-LAMP and RT-PCR because it has a shorter analysis times of 15–20 min at a lower temperature (37°C–42°C), it require less precision control and uses dried pellets, which are possible field advantages (Wang et al., 2020). Moreover, RPA primers have a strong tolerance for mismatch pairing (Daher et al., 2015). RT-RPA should be suitable for detecting viruses that exhibit enhanced mutation rates.
One out of five pairs of primers (HAV or norovirus) was selected to amplify the resulting single-stranded cDNA products and compare all three methods (RPA, conventional PCR, and qPCR). Although the one-step RT-RPA was quicker to set up and required less sampling handling, reducing pipetting errors. Two-step RT-RPA has many advantages over one-step RT-RPA. An RT reaction in a separate tube produces more stable cDNA than mRNA for later uses and greater flexibility to select the RT enzyme, thus improving reaction sensitivity (Euler et al., 2012). Since the same RT enzyme and conditions were used in this study, the detection limits of the three methods could be reliably estimated and compared.
Compared with conventional RT-PCR, simplex RT-RPA had greater sensitivity, possibly because RPA primers were used. The ideal length for PCR primers is around 18–25 bases. The shorter the primer, the more efficiently it will bind or anneal to the target. On the other hand, the longer the primer, the slower it will hybridize. The 35-bp long primer for the PCR reaction may affect the melting temperature that is critical for PCR. However, longer primers (28–35 bases) work better with highly variable viral genomes, such as norovirus. In addition, the relatively short amplicons produced in the RT-RPA reaction in this study may have increased the sensitivity of RT-RPA(Lobato and O’Sullivan, 2018). In RT-qPCR, development of standard curves and reaction efficiency is crucial for obtaining correct results. Although the standard regression lines showed relatively high correlation (R2 = 0.935 vs. 0.978) the slopes (−4.864 and −3.562) affected the reaction efficiency (60.543% and 90.885%), which may affect the detection limit of RT-qPCR by making it less sensitive than RT-RPA. In other words, performing RT-qPCR requires highly skilled personnel and precise control.
Multiplex PCR has been widely used to detect foodborne pathogens(Lee et al., 2018; Sheng et al., 2018). Compared with the simplex reaction, multiplex technology can minimizing the number of operational steps and reagents required to produce simultaneous multiple detections in a single reaction. The reduced cost, time, and labor make multiplexing an ideal strategy for pathogen identification. However, in multiplex PCRs, preferential amplification of one target sequence over another is a well-known phenomenon (Polz and Cavanaugh, 1998). The multiplex RT-PCR reaction using purified RNA performed amplification better for norovirus than for HAV, reflecting the competitive nature of the reaction. In this study, we optimized the reaction temperature, the concentration of template, and the primer concentration ratio of the norovirus target by reducing to half those of HAV. Thus, the reaction requires precisely controlled reaction components and conditions to obtain amplification efficiencies equivalent for all targets. However, optimizing multiplex PCRs can be tricky, resulting in low sensitivity or specificity (Polz and Cavanaugh, 1998). Similarly, the amplification efficiency of multiplex RT-LAMP was also limited by the many primers added to the reaction.
Unlike multiplex RT-PCR and RT-LAMP, multiplex RT-RPA has more flexibility regarding reaction conditions. RT-RPA has an added advantage in that tight temperature control is not required. In this study, duplex RT-RPA showed amplification efficiencies similar to those of HAV and norovirus with an equal amount of template targets and primers when using purified targets. However, in spiking tests, duplex RT-RPA showed superior amplification performance for norovirus than for HAV. Therefore, the concentrations of reaction components required adjustment, such as the concentration of Mg (Ac)2, and the ratios of the primers used. The majority of the developed multiplex RPA tests run in the temperature range of 37°C–41°C for 20 min. Long incubation times are not typically recommended because recombinase consumes all available ATP within 25 min. However, because many primers and targets are present in the same mixture, a longer amplification time may be required in the case of a multiplex reaction.
In the food matrix, enteric viruses are contaminants that threaten human health even though they are present in very low amounts. Before applying RT-RPA to on-site detection of foodborne viruses in under-equipped situations, it is also necessary to facilitate procedures for nucleic acid extraction. RT-RPA was shown to be tolerant to highly impure samples (Rajiuddin et al., 2020). Unlike RT-RPA, RT-PCR did not tolerate inhibitory compounds present in the oyster extract. Using our direct extraction method, RT-PCR obtained false-negative results. When using the detailed extraction protocol to obtain more purified targets (Beuret et al., 2003), RT-PCR could detect nucleic acids extracted from oysters but was less sensitive than RT-RPA. Comparing the two extraction methods showed that our extraction method saves 80% of the food processing time on average. Our extraction method will probably result in fewer steps and less dependence on the instrument, ultimately obtaining result within 2.5 hours.
The absence of virus-derived nucleic acids was a major limitation of this study. However, field evaluation of RT-RPA and sequence verification with DNA sequencing proved that RT-RPA had high sensitivity, specificity, and reliability. With the capability of direct detection of viruses in shellfish in a relatively short time and a reduced need for high analytical expertise, RT-RPA can be a promising alternative to RT-PCR-based or other isothermal methods in duplex testing for HAV and norovirus in shellfish. In this study, the sensitivities of all reactions, including RT-RPA, were much higher than those reported by other studies (Magro et al., 2017; Ming et al., 2015; Rames and Macdonald, 2019). It is probably due to the variation of spectrophotometry and cuvettes used to assess the concentration of RNA, which is the starting material. Moreover, end products were detected by agarose gel electrophoresis, with its sensitivity varying with the type of staining used. Using a fluorescence-labeled probe in the lateral flow assay to visualize the amplicons on the lateral flow strips increases the detection limits relative to RT-RPA alone (Hou et al., 2018; Xi et al., 2019). It is also more practical for on-site application than detecting end products by agarose gel electrophoresis. However,analysis of RT-RPA products carried out by agarose gel electrophoresis showed significant correlations between the band intensities of amplicons measured by the ImageJ program and the numbers of molecules. This result indicated that RT-RPA was suitable for quantification, but our finding was inconsistent with a previous repost in which RT-RPA produced non-linear curve (Ma et al., 2018).
This study demonstrated that the duplex RT-RPA assay was suitable for the primary screening of foodborne pathogens in shellfish products. The direct multiplex RPA assay was successfully applied to food samples (oysters) without sample processing, a significant advantage for field applications with limited resources. If used to analyze oyster farm samples by a trained professional, especially during primary production, which is a critical stage of food processing, the technique can minimize the risk of infection to consumers, thereby improving food safety.
ACKNOWLEDGEMENTS
We are grateful to Professor Dr. Somsak Pantuwatana, and Professor Dr.Wanpen Chaicumpa, Faculty of Medicine, Siriraj Hospital, Mahidol University, for valuable advice and great support.
AUTHOR CONTRIBUTIONS
Uraiwan Intamaso designed the experiments and wrote the manuscript. Palatip Chutoam performed the statistical analysis and data visualization. Supannee Lethochavalit collected samples. Suthasinee Jinda conducted the experiments. All authors have read and approved of the final manuscript.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
Aebischer, A., Wernike, K., Hoffmann, B., and Beer, M. 2014. Rapid genome detection of Schmallenberg virus and bovine viral diarrhea virus by use of isothermal amplification methods and high-speed real-time reverse transcriptase PCR. Journal of Clinical Microbiology. 52: 1883–1892.
Barameechai, K., Sa-Nguanmoo, P., Suwannakarn, K., Thongmee, C., Payungporn, S., Chongsrisawat, V., Theamboonlers, A., and Poovorawan, Y. 2008. Molecular characterisation of the hepatitis A virus circulating in the 2001-2005 outbreaks in Thailand. Annals of Tropical Medicine and Parasitology. 102: 247–257.
Beuret, C., Baumgartner, A., and Schluep, J. 2003. Virus-contaminated oysters: A three-month monitoring of oysters imported to Switzerland. Applied and Environmental Microbiology. 69: 2292–2297.
Carter, M.J. 2005. Enterically infecting viruses: Pathogenicity, transmission and significance for food and waterborne infection. Journal of Applied Microbiology. 98: 1354–1380.
Centers for Disease Control and Prevention (CDC). 2021. Norovirus Worldwide| CDC. https://www.cdc.gov/norovirus/trends-outbreaks/worldwide.html
Chuchaona, W., Chansaenroj, J., Wanlapakorn, N., Vongpunsawad, S., and Poovorawan, Y. 2019. Recombinant GII.Pe-GII.4 Norovirus, Thailand, 2017-2018. Emerging Infectious Diseases. 25: 1612–1614.
Crannell, Z., Castellanos-Gonzalez, A., Nair, G., Mejia, R., White, A.C., and Richards-Kortum, R. 2016. Multiplexed recombinase polymerase amplification assay to detect intestinal protozoa. Analytical Chemistry. 88: 1610–1616.
Daher, R.K., Stewart, G., Boissinot, M., and Bergeron, M.G. 2016. Recombinase polymerase amplification for diagnostic applications. Clinical Chemistry. 62: 947–958.
Daher, R.K., Stewart, G., Boissinot, M., Boudreau, D.K., and Bergeron, M.G. 2015. Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Molecular and Cellular Probes. 29: 116–121.
Euler, M., Wang, Y., Nentwich, O., Piepenburg, O., Hufert, F.T., and Weidmann, M. 2012. Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus. Journal of Clinical Virology. 54: 308–312.
Fang, P.-Y., Bowman, J.C., Ramos, L.M.G., Hsiao, C., and Williams, L.D. 2018. RNA: Packaged and protected by VLPs. RSC Advances. 8: 21399–21406.
Han, Y., Wang, J., Zhang, S., Wang, J., Qin, C., Han, Y., and Xu, X. 2020. Rapid detection of norovirus genogroup II in clinical and environmental samples using recombinase polymerase amplification. Analytical Biochemistry. 605: 113834.
Hou, P., Zhao, G., Wang, H., He, C., Huan, Y., and He, H. 2018. Development of a recombinase polymerase amplification combined with lateral-flow dipstick assay for detection of bovine ephemeral fever virus. Molecular and Cellular Probes. 38: 31–37.
Kibbe, W. A. 2007. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Research. 35(Web Server issue): W43-46.
Lee, S.-Y., Kim, M.-J., Kim, H.-J., Jeong, K.C., and Kim, H.-Y. 2018. Simultaneous detection of four foodborne viruses in food samples using a one-step multiplex reverse transcription PCR. Journal of Microbiology and Biotechnology. 28: 210–217.
Lemon, S.M., Ott, J.J., Van Damme, P., and Shouval, D. 2017. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. Journal of Hepatology. 278: 167-184
Li, G., Wu, M., Li, J., Cai, W., Xie, Y., Si, G., Xiao, L., Cong, F., and He, D. 2021. Rapid detection of porcine deltacoronavirus and porcine epidemic diarrhea virus using the duplex recombinase polymerase amplification method. Journal of Virological Methods. 292: 114096.
Li, J., Macdonald, J., and Stetten, F. von. 2018. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst. 144: 31–67.
Lobato, I.M., and O’Sullivan, C.K. 2018. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends in Analytical Chemistry. 98: 19–35.
Ma, B., Li, J., Chen, K., Yu, X., Sun, C., and Zhang, M. 2020. Multiplex recombinase polymerase amplification assay for the simultaneous detection of three foodborne pathogens in seafood. Foods. 9: 278.
Ma, L., Zeng, F., Cong, F., Huang, B., Zhu, Y., Wu, M., Xu, F., Yuan, W., Huang, R., and Guo, P. 2018. Development and evaluation of a broadly reactive reverse transcription recombinase polymerase amplification assay for rapid detection of murine norovirus. BMC Veterinary Research. 14: 399.
Magro, L., Jacquelin, B., Escadafal, C., Garneret, P., Kwasiborski, A., Manuguerra, J.-C., Monti, F., Sakuntabhai, A., Vanhomwegen, J., Lafaye, P., and Tabeling, P. 2017. Paper-based RNA detection and multiplexed analysis for Ebola virus diagnostics. Scientific Reports. 7: 1347.
Ming, K., Kim, J., Biondi, M.J., Syed, A., Chen, K., Lam, A., Ostrowski, M., Rebbapragada, A., Feld, J.J., and Chan, W.C.W. 2015. Integrated quantum dot barcode smartphone optical device for wireless multiplexed diagnosis of infected patients. ACS Nano. 9(3): 3060–3074.
Moore, M.D., and Jaykus, L.-A. 2017. Development of a recombinase polymerase amplification assay for detection of epidemic human noroviruses. Scientific Reports. 7: 40244.
Piepenburg, O., Williams, C.H., Stemple, D.L., and Armes, N.A. 2006. DNA detection using recombination proteins. PLoS Biology. 4: e204.
Pintó, R.M., Costafreda, M.I., and Bosch, A. 2009. Risk assessment in shellfish-borne outbreaks of hepatitis A. Applied and Environmental Microbiology. 75: 7350–7355.
Polz, M.F., and Cavanaugh, C.M. 1998. Bias in Template-to-product ratios in multitemplate PCR. Applied and Environmental Microbiology. 64: 3724–3730.
Rajiuddin, S.M., Midgley, S.E., Jensen, T., Müller, L., and Schultz, A.C. 2020. Application of an optimized direct lysis method for viral RNA extraction linking contaminated dates to infection with hepatitis a virus. Frontiers in Microbiology. 11: 516445.
Rames, E.K. and Macdonald, J. 2019. Rapid assessment of viral water quality using a novel recombinase polymerase amplification test for human adenovirus. Applied Microbiology and Biotechnology. 103: 8115–8125.
Sheng, J., Tao, T., Zhu, X., Bie, X., Lv, F., Zhao, H., and Lu, Z. 2018. Multiplex PCR detection method for milk based on novel primers specific for Listeria monocytogenes 1/2a serotype. Food Control. https://agris.fao.org/agris-search/search.do?recordID=US201800124956
Studier, F.W. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification. 41: 207–234.
Tran, D.H., Tran, H.T., Pham, T.N.M., and Phung, H.T.T. 2022. Direct multiplex recombinase polymerase amplification for rapid detection of Staphylococcus aureus and Pseudomonas aeruginosa in food. Molecular Biology Research Communications. 11: 1–10.
Wang, Y., Chen, R., Nie, X., Zhong, Z., Li, C., Li, K., Huang, W., Fu, X., Liu, J., and Nie, B. 2020. Rapid and sensitive detection of potato virus Y by isothermal reverse transcription-recombinase polymerase amplification assay in potato. Molecular and Cellular Probes. 50: 101505.
World Health Organization. 2022a. Food safety. https://www.who.int/news-room/fact-sheets/detail/food-safety
World Health Organization. 2022b. Hepatitis A. https://www.who.int/news-room/fact-sheets/detail/hepatitis-a
Xi, Y., Xu, C.-Z., Xie, Z.-Z., Zhu, D.-L., and Dong, J.-M. 2019. Rapid and visual detection of dengue virus using recombinase polymerase amplification method combined with lateral flow dipstick. Molecular and Cellular Probes. 46: 101413.
Yoda, T., Suzuki, Y., Yamazaki, K., Sakon, N., Kanki, M., Aoyama, I., and Tsukamoto, T. 2007. Evaluation and application of reverse transcription loop-mediated isothermal amplification for detection of noroviruses. Journal of Medical Virology. 79: 326–334.
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand.
Palatip Chutoam1, Suthasinee Jinda1, Supannee Lethochavalit2, Uraiwan Intamaso1, *
1Faculty of Allied Health Sciences, Burapha University, 169 Long-Hard Bangsaen Rd., Chonburi 20131, Thailand.
2 Institute of Marine Science, Burapha University, 169 Long-Hard Bangsaen Rd., Chonburi 20131, Thailand.
*Corresponding author: Uraiwan Intamaso, E-mail: uraiwani@go.buu.ac.th
Total Article Views
Editor: Waraporn Boonchieng,
Chiang Mai University, Thailand
Article history:
Received: May 5, 2022;
Revised: October 29, 2022;
Accepted: November 4, 2022;
Published online: November 15, 2022