 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Design of New Chlorochalcone Derivatives as Potential Breast Anticancer Compound Based on QSAR Analysis and Molecular Docking Study
Anita Dwi Puspitasari, Harno Dwi Pranowo, Endang Astuti and Tutik Dwi Wahyuningsih*Published Date : 2021-05-11
DOI : https://doi.org/10.12982/CMUJNS.2021.070
Journal Issues : Number 3, July-September 2021
Abstract Quantitative structure-activity relationships (QSAR) proposes a model that relates the biological activities of drugs to their chemical structures, and the interaction between the drug and its target enzyme is revealed by molecular docking research. These studies were conducted on chalcone to produce a model that could design highly potent breast anticancer MCF7 cells. The compounds were optimized using ab initio using a basis set 6-31G, then their descriptors calculated using this method. Genetic Function Algorithm (GFA) was used to select descriptors and build the model. One of the six models generated was found to be the best with internal and external squared correlation coefficient (R2) of 0.743 and 0.744, respectively, adjusted squared correlation coefficient (adjusted R2) of 0.700, Standard estimate of error (SEE) of 0.198, Fcalc/Ftable of 6.423, and Predicted residual sum of squares (PRESS) of 1.177. The QSAR equation is pIC50 = 3.869 + (1.427 x qC1) + (4. 027 x qC10) + (0.856 x qC15) - (35.900 x ELUMO) + (0.208 x Log P). Hence, it can predict the breast anticancer activities of new chlorochalcones A-F. The compound with the best prediction was chlorochalcone A with pIC50 2.65 and IC50 value of 2.26 μM. The chlorochalcones A-F were able to bind to the main amino acid residues, namely Arg120 and Tyr355, on the active site of the COX-2 enzyme. These results could serve as a model for designing novel chlorochalcone as inhibitors of COX-2 with higher breast anticancer activities.
Keywords: Chlorochalcone, COX-2, QSAR, MCF-7, Molecular docking
Funding: The authors are grateful for the financial support provided by The Ministry of Research, Technology, and Higher Education of The Republic of Indonesia.
Citation: Puspitasari, A.D., Pranowo, H.D., Astuti, E., and Wahyuningsih, T.D. 2021. Design of new chlorochalcone derivatives as potential breast anticancer compound based on QSAR analysis and molecular docking study. CMUJ. Nat. Sci. 20(3): e2021070
INTRODUCTION
Globally, cancer is a non-communicable disease that causes the most deaths after stroke and hypertension, with a death rate of around 9,6 million in 2018 (Wild et al., 2020). Based on the top 10 cancer ranked in women, breast cancer is in the first rank with an incidence rate of 42.1 per 1000 population (Globocan, 2018). High cytotoxicity and drug resistance are key breast cancer treatment issues, and no clinically active substances are known to work selectively on tumor cells (Ivkovic et al., 2013). The process of breast cancer is regulated by various pathways involving different enzymes. The enzyme that plays a role in the synthesis of prostaglandins associated with breast cancer development is COX-2, which was found to be overexpressed in MCF7 breast cancer cells. Inhibition of this enzyme can potentially develop anticancer drugs (Krishnamachary et al., 2017).
Chalcone, as shown in Figure 1, is reported to have many biological activities, including anti-inflammatory, antiviral, anti-protozoa, insecticidal, and anticancer activity (Dimmock et al., 2000; Yadav et al., 2011). As a compound with anticancer activity, chalcone shows anti-proliferation, induction of apoptosis, anti-metastasis, anti-angiogenesis, DNA and mitochondrial damage, and tubulin inhibition (Das and Manna, 2016). By adding various functional groups such as aryls, halogens, hydroxyl, carboxylate groups, and benzene, chalcone biological activity may change due to their interactions with different molecular targets (Jandial et al., 2014).
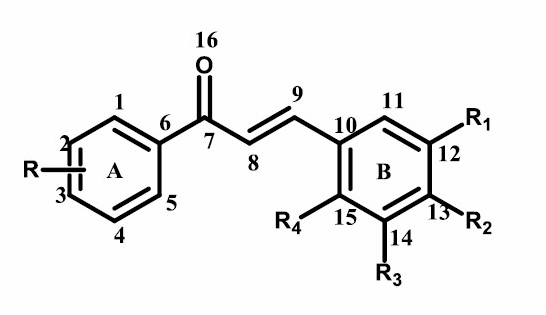
Figure 1. Structure of Chalcone.
Quantitative structure-activity relationship (QSAR) is a method in designing new drugs through a ligand-based approach, whereas molecular docking is a method used to determine the binding strength between the active site residue and the ligand. The QSAR of chalcones with an anticancer activity has been done with HT-29 human colon adenocarcinoma cell lines (Rybka et al., 2014). Meanwhile, molecular docking of chalcone derivatives that have been done was against cyclin-dependent kinase CDK7 (Khanapure et al., 2018), α-β tubulin (Pingaew et al., 2014), α-estrogen receptors (Gurrapu et al., 2020), and EGFR (Suma et al., 2019). The combination of QSAR analysis with molecular docking for designing the chalcone derivatives as anticancer towards MCF7 cancer cell lines had not been reported.
This paper looks at the QSAR of chalcones against human breast cancer cells (MCF7) based on 48 chalcone derivatives, calculated under the same conditions. In theory, this group of chalcones will give reliable QSAR parameters for determining the activity of certain newly synthesized chalcone derivatives as anticancer of MCF7 cell lines. The newly chalcone derivatives were subsequently used for molecular docking analysis with COX-2 as a target for breast anticancer activity.
MATERIALS AND METHODS
Materials
A series of chalcone derivatives consist of 48 compounds along with their cytotoxic activity against MCF7 cells from the research of Suma et al., 2019; Syam et al., 2012; Raghavan et al., 2015; Ivkovic et al., 2013; and Shenvi et al., 2013 were used for the QSAR analysis. The proposed compound obtained from QSAR analysis and the crystal structure of the Cyclooxygenase-2 enzyme with PDB ID: 4COX and 6COX were used for molecular docking.
Data set preparation
A total of 48 chalcone derivatives (Table 1) were selected from the recently published literature, i.e., Suma et al., 2019; Syam et al., 2012; Raghavan et al., 2015; Ivkovic et al., 2013; and Shenvi et al., 2013. All of their activities towards MCF7 cell lines have been reported using the same biological assay method. All compounds were evaluated for their cytotoxic activity by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl diphenyltetrazolium bromide (MTT ASSAY) based on the reduction of mitochondria from yellow MTT tetrazolium to very colorful blue formazan product. The selected data set compounds were determined based on the following criteria, having the basic structure of chalcone (1,3-diaryl-2-propen-1-ones) and the Inhibition concentration (IC50) values are less than 200 μM. The IC50 (μM) value was converted to molar concentration then converted to the corresponding pIC50 (-log IC50) (M). The range of pIC50 values is between 0.74 - 3.1 in the data set, divided into 2 sets, namely the training set and the test set.
Table 1. pIC50 value of chalcone derivative compounds against MCF7 cells.
|
No |
R |
R1 |
R2 |
R3 |
R4 |
pIC50 |
|
1a |
3-Cl |
H |
OCH3 |
H |
H |
1.00 |
|
2a |
3-Cl |
OCH3 |
OCH3 |
H |
H |
3.10 |
|
3a |
3-Cl |
OCH3 |
OH |
H |
H |
2.12 |
|
4a |
H |
H |
H |
H |
H |
2.16 |
|
5b |
3-CH3 |
H |
H |
H |
H |
1.87 |
|
6a |
3-OCH3 |
H |
H |
H |
H |
1.71 |
|
7b |
H |
H |
H |
H |
Cl |
2.10 |
|
8b |
3-OH |
H |
H |
H |
Cl |
2.00 |
|
9a |
H |
CH3 |
H |
H |
H |
2.03 |
|
10a |
3-OH |
CH3 |
H |
H |
H |
2.08 |
|
11a |
3-CH3 |
CH3 |
H |
H |
H |
1.99 |
|
12a |
3-OCH3 |
CH3 |
H |
H |
H |
2.02 |
|
13a |
H |
H |
OCH3 |
H |
H |
1.93 |
|
14a |
3-CH3 |
H |
OCH3 |
H |
H |
1.49 |
|
15a |
3-OCH3 |
H |
OCH3 |
H |
H |
1.38 |
|
16a |
H |
H |
N(CH3)2 |
H |
H |
1.01 |
|
17b |
3-CH3 |
H |
N(CH3)2 |
H |
H |
1.24 |
|
18a |
H |
H |
H |
H |
OH |
2.03 |
|
19a |
3-CH3 |
H |
H |
H |
OH |
2.16 |
|
20a |
3-OCH3 |
H |
H |
H |
OH |
2.15 |
|
21a |
H |
Cl |
H |
H |
H |
2.28 |
|
22a |
5-OH |
H |
H |
H |
CF3 |
1.72 |
|
23a |
5-OH |
H |
H |
H |
CH3 |
1.42 |
|
24a |
5-OH |
H |
H |
H |
F |
1.84 |
|
25b |
5-OH |
H |
H |
H |
Cl |
1.53 |
|
26a |
5-OH |
H |
H |
F |
H |
1.58 |
|
27a |
5-OH |
H |
H |
CH3 |
H |
1.47 |
|
28a |
3-Cl |
OCH3 |
OCH3 |
H |
OCH3 |
1.53 |
|
29a |
3-OCH3 |
OCH3 |
OCH3 |
H |
OCH3 |
1.47 |
|
30b |
3-CH3 |
OCH3 |
OCH3 |
H |
OCH3 |
1.48 |
|
31a |
3,5-OH |
OCH3 |
OCH3 |
H |
OCH3 |
1.47 |
|
32a |
3-F |
OCH3 |
OCH3 |
H |
OCH3 |
1.52 |
|
Table 1. Continued. |
||||||
|
No |
R |
R1 |
R2 |
R3 |
R4 |
pIC50 |
|
33a |
2-NO2, 3-Cl |
OCH3 |
OCH3 |
H |
OCH3 |
1.79 |
|
34a |
2-OCH3, 3-OH |
OCH3 |
OCH3 |
H |
OCH3 |
1.02 |
|
35a |
1, 3, 5-OCH3 |
OCH3 |
OCH3 |
H |
OCH3 |
1.29 |
|
36b |
3-Br |
OCH3 |
OCH3 |
H |
OCH3 |
1.74 |
|
37a |
3-OH |
OCH3 |
OCH3 |
H |
OCH3 |
0.74 |
|
38a |
H |
OCH3 |
OH |
H |
H |
1.44 |
|
39a |
3-OCH3 |
OCH3 |
OH |
H |
H |
1.13 |
|
40b |
3-F |
OCH3 |
OH |
H |
H |
1.92 |
|
41a |
3-NO2 |
OCH3 |
OH |
H |
H |
2.07 |
|
42a |
3,5-Cl |
OCH3 |
OH |
H |
H |
1.89 |
|
43a |
H |
OH |
OCH3 |
H |
H |
1.34 |
|
44a |
3-OCH3 |
OH |
OCH3 |
H |
H |
1.13 |
|
45a |
3-Cl |
OH |
OCH3 |
H |
H |
1.77 |
|
46a |
3,5-Cl |
OH |
OCH3 |
H |
H |
2.00 |
|
47a |
H |
OCH3 |
OCH3 |
H |
H |
1.42 |
|
48a |
3-OH |
OCH3 |
OCH3 |
H |
H |
1.39 |
Note: a Training-set; b Test-set
Computational methods
The best computational method was selected by predicting the 1H-NMR chemical shift, which is calculated using the semi-empirical method (AM1, PM3, and PM6), the ab initio method (HF 3-21G, HF 6-31G, and HF 6-311G) and the DFT method with the functional hybrid B3LYP basis set 3-21G, 6-31G, and 6-311G. The result was then compared with the experimental data of 1H-NMR (Table 2).
All quantum mechanical calculations were executed using Gaussian 09 (Frisch et al., 2009). Correlation models were evaluated by multiple linear regression analysis using Build-QSAR (De Oliveira and Gaudio, 2000).
Electronic and molecular descriptor calculations
The calculation of electronic and molecular descriptors was carried out by optimizing the molecular geometry of the 48 chalcones derivatives. Geometry optimization with the best computational method was performed using Gaussian® 09W software.
Model validation
The resulting QSAR model was selected based on statistical parameters such as the correlation coefficient (R), the coefficient of determination (R2), the standard estimation error (SEE), Fcal/Ftab values, and the predicted residual error sum of squares (PRESS) (Veerasamy et al., 2014).
Design of new compounds
The new chalcone derivatives were designed by considering the descriptors that affect the resulted QSAR equation and based on the correlation of the substituent with the pIC50 value. The new compounds were simulated using the molecular docking method to determine their interaction with the COX-2 enzyme.
Material preparation for molecular docking
Three-dimension (3D) structures of cyclooxygenase-2 (COX-2) enzymes with PDB ID: 4COX and 6COX were downloaded from the Protein Data Bank database (www. rcsb. org) 4COX and 6COX are complex COX-2 enzymes with native ligand indomethacin and SC-558, a selective COX-2 inhibitor. A series of chlorochalcone derivatives (compound A-F) were used as experimental ligands.
The 3D structures of COX-2 were prepared with YASARA (http://www.yasara.org) in the standard-setting. Only an A-chain of the protein extracted from the PDB file. At this stage, the water molecule and the reference ligand are removed, and the hydrogen atoms were added. The results were saved in the .mol2 format and would be a virtual target for docking simulation. The native ligand Indomethacin and SC-558 were prepared with Marvin sketch by configuring them into two dimensions (2D) formats. They were protonated at pKa 7.4, and ligand conformations were performed. The ten conformers form of indomethacin and SC-558 ligands were saved in the .mol2 format for the next docking process.
Chlorochalcone ligands as input structure were drawn using Marvin Sketch software. The preparation for these ligands was performed in the same way as the native ligands, and the conformers were saved in the .mol2 format for the docking process.
Molecular docking process
All the prepared ligands were docking molecularly using PLANTS (Protein-Ligand Ant System) (Korb et al., 2009). Protocol validation was carried out to determine the root mean square deviation value (RMSD). Validation using the YASARA program (Analyze> RMSD> Molecule) with inserts specific ligands and receptors with the .mol2 format. A protocol was accepted when the RMSD heavy atoms are less than 2.0 Å (Sokalingam et al., 2018). The docking results can be seen in the output in notepad format. The determination of docking result complex conformation is done by choosing the selected conformation with the lowest CHEMPLP score. Visualized docking results using Discovery Studio software.
RESULTS
Computational methods
The results of 1H-NMR chemical shift calculation were listed in Table 2. The compound (2), as shown in Figure 2, was used as a parameter because it has the best activity against breast cancer cell line MCF7.
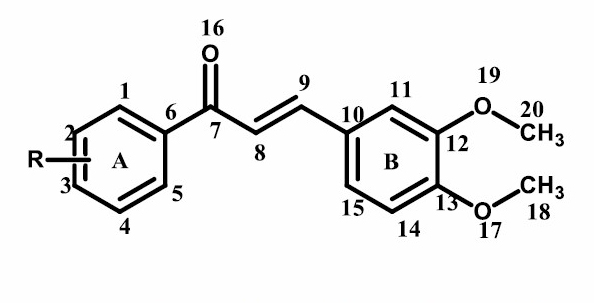
Figure 2. (E)-1-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one.
Table 2. Differences in chemical shift of 1H-NMR with experimental and computational methods (δ, ppm).
|
Hydrogen atoms |
δExp |
The semi-empirical method |
The ab initio method |
The DFT method with The functional hybrid B3LYP |
||||||
|
δAM1 |
δPM3 |
δPM6 |
δHF 3-21G |
δHF 3-21G |
δHF 3-21G |
δDFT 3-21G |
δDFT 3-21G |
δDFT 3-21G |
||
|
H18 |
3.93 |
4.36 |
3.81 |
3.89 |
3.89 |
3.81 |
3.77 |
3.95 |
3.85 |
3.95 |
|
H20 |
4.94 |
4.60 |
4.19 |
4.56 |
4.56 |
4.56 |
4.28 |
4.20 |
5.06 |
4.19 |
|
H14 |
6.89 |
7.24 |
7.21 |
6.63 |
6.63 |
6.71 |
6.68 |
7.00 |
7.02 |
7.09 |
|
H11 |
7.14 |
7.77 |
7.74 |
7.20 |
7.20 |
7.26 |
7.22 |
7.50 |
7.52 |
7.58 |
|
H15 |
7.25 |
7.98 |
8.66 |
7.66 |
7.66 |
7.66 |
7.63 |
7.98 |
7.97 |
8.04 |
|
H8 |
7.34 |
7.88 |
9.08 |
7.55 |
7.55 |
7.41 |
7.39 |
7.93 |
7.88 |
7.96 |
|
H2 |
7.46 |
7.92 |
7.83 |
7.37 |
7.37 |
7.48 |
7.48 |
7.68 |
7.71 |
7.78 |
|
H4 |
7.46 |
7.78 |
7.81 |
7.35 |
7.35 |
7.38 |
7.33 |
7.66 |
7.63 |
7.69 |
|
H9 |
7.76 |
8.48 |
8.02 |
8.06 |
8.06 |
8.00 |
7.95 |
8.46 |
8.33 |
8.42 |
|
H1 |
7.95 |
8.42 |
8.56 |
8.6 |
8.60 |
8.30 |
8.28 |
9.05 |
8.83 |
8.92 |
|
H5 |
7.95 |
8.10 |
8.81 |
7.88 |
7.88 |
7.85 |
7.82 |
8.18 |
8.15 |
8.21 |
|
R2 |
0.96 |
0.91 |
0.97 |
0.97 |
0.98 |
0.98 |
0.95 |
0.98 |
0.96 |
|
|
PRESS |
2.72 |
7.49 |
1.43 |
0.97 |
0.58 |
0,84 |
3.19 |
2.22 |
3.40 |
|
Electronic and molecular descriptor calculations
The electronic descriptors obtained were the net atomic charge, HOMO, and LUMO energy, while the molecular descriptors were molecular surface area, molecular volume, logarithm of the partition coefficient (log P), molar refractivity, polarizability, and molecular mass. The symbol and unit of the descriptor used for constructing the QSAR models were shown in Table 3.
Table 3. Electronic and molecular descriptors used to construct the QSAR equation.
|
Descriptors |
Symbol |
Unit |
|
Net atomic charge for C |
qC |
Coulomb |
|
Net atomic charge for O |
qO |
Coulomb |
|
HOMO energies |
E_HOMO |
eV |
|
LUMO energies |
E_LUMO |
eV |
|
Molecular surface area |
LA |
Å2 |
|
Molecular volume |
VOL |
Å3 |
|
Molar refractivity |
MR |
Å-3 |
|
Logarithm of the octanol/water partition coefficient |
logP |
- |
|
Molecular mass |
MW |
Amu |
|
Polarizability |
POL |
Å3 |
Model Validation
The best QSAR equation model was determined from the results of QSAR analysis with electronic and molecular parameters. The best QSAR models' selection was based on statistical parameters such as R2 value (coefficient correlation), adjusted R2, Fcal/Ftab, and PRESS (Predicted Residual Sum of Square). Based on this data, six models were developed, as listed in Table 4.
Table 4. QSAR models and statistical parameters.
|
Model |
Descriptors |
R |
R2 |
R2adj |
SEE |
F |
Fcal/Ftab |
PRESS |
|||||||||
|
1 |
qC1, qC15, E_LUMO, Log P |
0.85 |
0.72 |
0.69 |
0.20 |
20.29 |
6.94 |
1.27 |
|
||||||||
|
2 |
qC1, qC10, qC15, E_LUMO, Log P |
0.86 |
0.74 |
0.70 |
0.20 |
17.35 |
6.42 |
1.18 |
|
||||||||
|
3 |
qC1, qC8, qC15, qC17, E_LUMO, Log P |
0.88 |
0.78 |
0.73 |
0.19 |
12.86 |
5.03 |
1.01 |
|
||||||||
|
4 |
qC1, qC8, qC11, qC15, qC17, E_LUMO, Log P |
0.88 |
0.78 |
0.72 |
0.19 |
14.28 |
5.81 |
1.00 |
|
||||||||
|
5 |
qC1,qC2, qC8, qC11, qC15, qC17, E_LUMO, Log P |
0.88 |
0.78 |
0.72 |
0.19 |
12.05 |
5.05 |
1.00 |
|
||||||||
|
6 |
qC1, qC2, qC5, qC8, qC11, q15, qC17, E_LUMO, Log P |
0.88 |
0.78 |
0.71 |
0.20 |
10.32 |
4.42 |
1.00 |
|
||||||||
The six models were tested for external validity to determine the models’ ability to predict anticancer activity in the test set. External validation was carried out to test the six QSAR models' ability that fulfills statistical criteria in predicting anticancer activity outside the training set. The statistical criteria in performing external validation used were the predicted R2 value and PRESS, as listed in Table 5.
Table 5. Comparison of the experimental pIC50 with the predicted pIC50 in the test set.
|
Test-Set |
pIC50 Experiment |
pIC50 Predictions |
|||||||||||||
|
Model 1 |
Model 2 |
Model 3 |
Model 4 |
Model 5 |
Model 6 |
||||||||||
|
5 |
1.87 |
1.99 |
1.96 |
2.07 |
2.07 |
2.07 |
2.07 |
|
|||||||
|
7 |
2.10 |
1.92 |
2.09 |
1.87 |
1.86 |
1.86 |
1.87 |
|
|||||||
|
8 |
2.00 |
1.66 |
1.85 |
1.58 |
1.57 |
1.57 |
1.59 |
|
|||||||
|
17 |
1.24 |
1.34 |
1.23 |
1.01 |
0.99 |
0.99 |
1.00 |
|
|||||||
|
25 |
1.53 |
1.50 |
1.73 |
1.28 |
1.27 |
1.27 |
1.27 |
|
|||||||
|
30 |
1.48 |
1.46 |
1.47 |
1.39 |
1.39 |
1.40 |
1.40 |
|
|||||||
|
36 |
1.74 |
1.86 |
1.83 |
1.75 |
1.76 |
1.76 |
1.76 |
|
|||||||
|
40 |
1.92 |
1.65 |
1.64 |
1.77 |
1.77 |
1.77 |
1.78 |
|
|||||||
|
PRESS |
0.26 |
0.16 |
0.42 |
0.44 |
0.44 |
0.41 |
|||||||||
|
R2 prediction |
0.62 |
0.74 |
0.70 |
0.69 |
0.69 |
0.71 |
|||||||||
A graphical plot between the observed and calculated values of pIC50 for all compounds in the data set was presented in Figure 3.
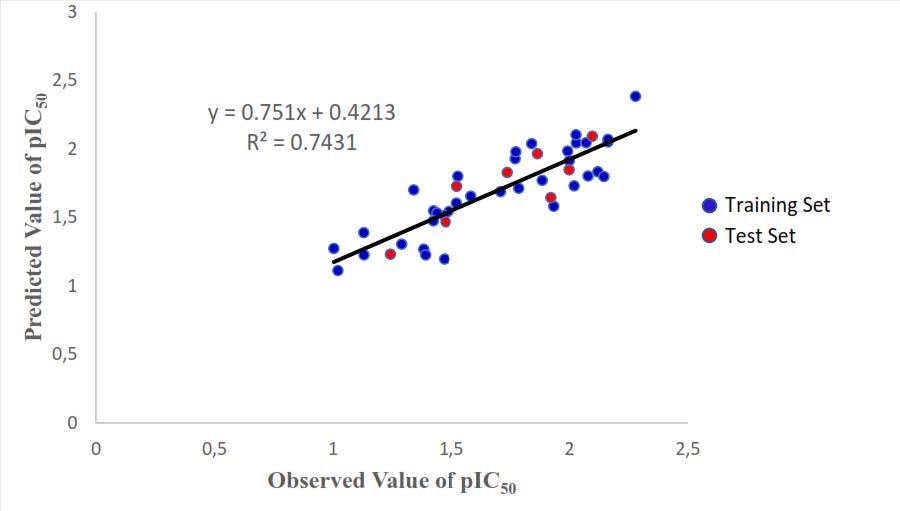
Figure 3. A Plot of observed and predicted pIC50 values of the whole set.
Design of new compound
We have designed the chalcone derivatives by replacing the R substituent based on model 2, to increase the net atomic charge of C1, C10, C15, and log P to minimize LUMO energy. The result was shown in Table 6.
Table 6. Newly designed chlorochalcone derivatives and their prediction of activity against MCF-7 cells calculated using the best QSAR.
Docking method validation
The native ligand was docked into cyclooxygenase-2 (COX-2) enzymes to visualize the binding pose of protein. The Indomethacin ligand (IMN) was docked into COX-2 enzymes (PDB ID: 4COX) and SC-558 into COX-2 enzymes (PDB ID: 6COX). The comparison between the ligand conformation Indomethacin and SC-558 from the x-ray crystal structure with re-docking calculation results can be seen in Figure 4.
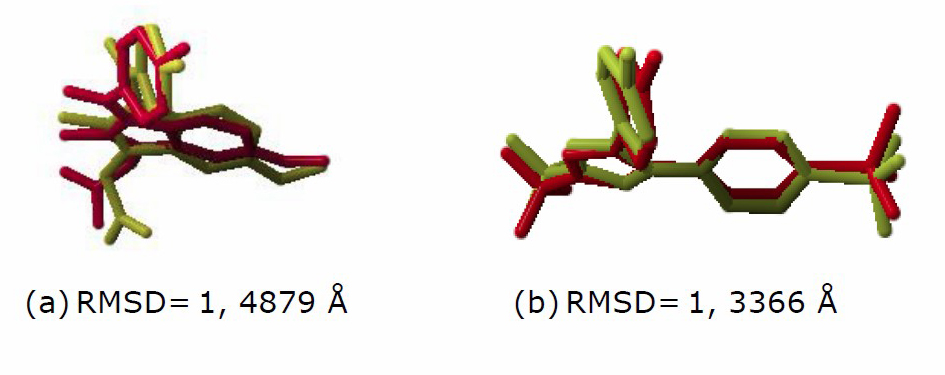
Figure 4. Comparison of conformation between the native ligand of the X-ray crystal structure (yellow) to the docking result (red) and its RMSD values
(a) Indomethacin (4COX), (b) SC-558 (6COX).
Docking study of newly design chlorochalcone compounds
The docking results of native ligand and newly chlorochalcone compounds against COX-2 enzyme were shown in Table 7.
Table 7. Results of Newly Chlorochalcone docking towards enzyme COX-2 (4COX and 6COX) and hydrogen bond positions in the receptor–compound interaction.
|
Compound |
Free Binding Energy |
Hydrogen Bond (n) |
|||
|
4COX |
6COX |
4COX |
6COX |
||
|
SC-558 |
- |
-71.3196 |
- |
Arg120, Gln192, Leu352 |
|
|
Indomethacin |
-75.4911 |
- |
Tyr355 (2) |
- |
|
|
A |
-92.2053 |
-79.4650 |
- |
Tyr355 |
|
|
B |
-91.2850 |
-83.9575 |
- |
Tyr385 |
|
|
C |
-87.9721 |
-82.3396 |
- |
- |
|
|
D |
-91.2435 |
-79.5004 |
- |
Arg120 |
|
|
E |
-87.6766 |
-80.9845 |
- |
- |
|
|
F |
-89.7247 |
-84.9019 |
Tyr355 |
Met522 |
|
Notes: n, number in parentheses indicates more than one H-bond in the amino acid residue.
“–” indicates no H-bond formed in the amino acid residue.
The visualization results with the Discovery Studio program from the interaction between native ligand and Chalcone A with the active site of the COX-2 enzyme were presented in Figures 5 and 6.
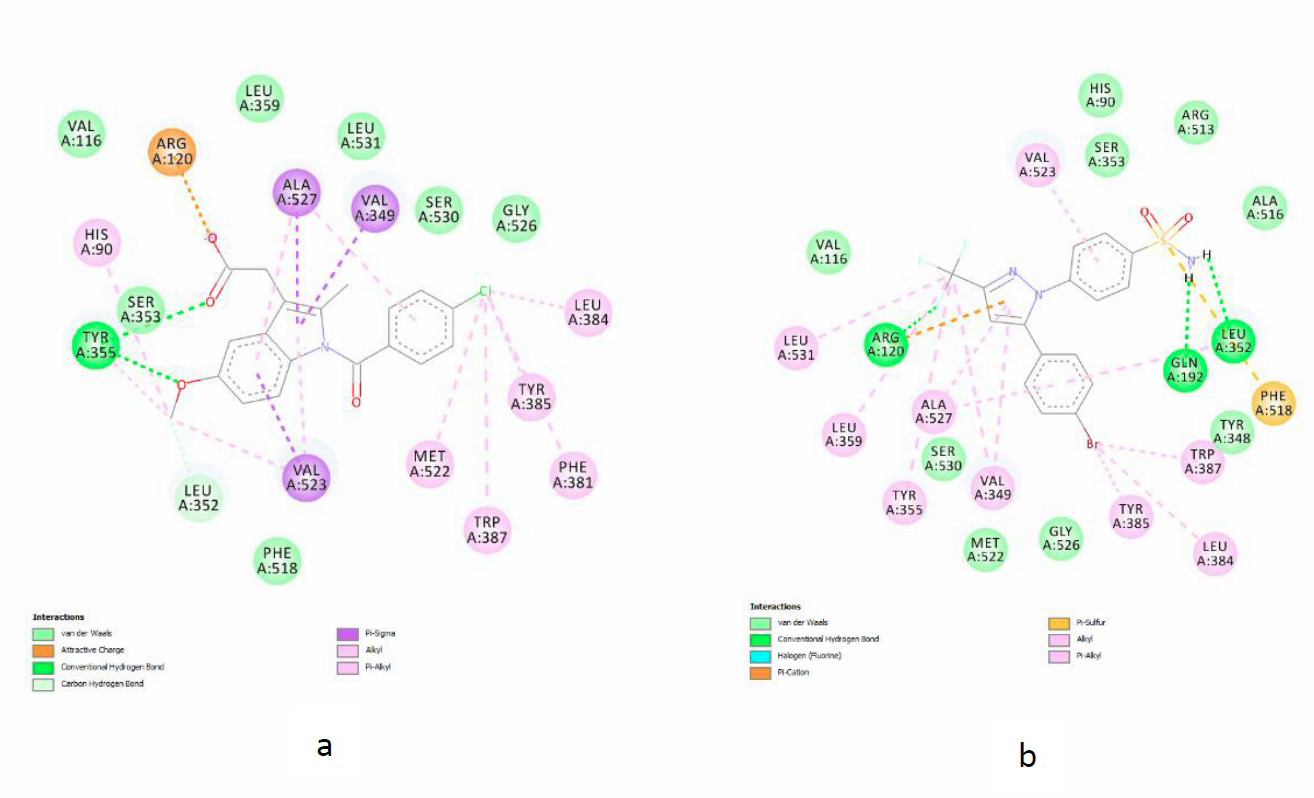
Figure 5. Interaction of native ligand on the active site of COX-2 enzymes (a) Indomethacin (4COX), (b) SC-558 (6COX).
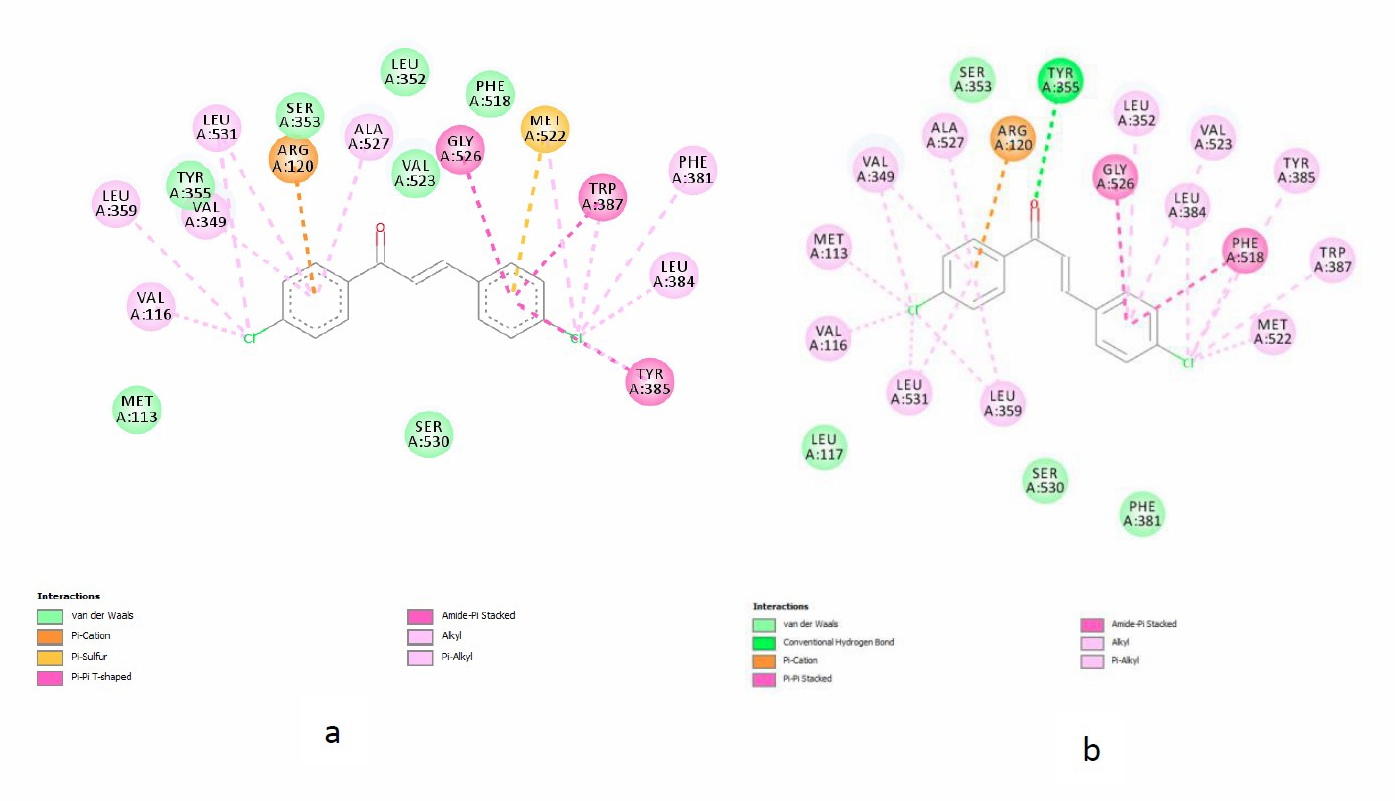
Figure 6. Interaction of chalcone A on the active site of COX-2 enzymes (a) 4COX, (b) 6COX.
DISCUSSIONS
Computational methods
The process of geometry optimization is the most important step in computational chemistry before analyzing the test compound because computational chemistry calculations will be based on molecular conformations (Onlu and Sacan, 2017). The 1H-NMR chemical shift value was used to validate the computational method in optimizing the geometry of 48 chalcone-derived compounds, which will be used to calculate electronic and molecular descriptors. This is because the chemical shift is related to the atom's partial charge, which will determine the compound’s physical and chemical properties. The value of the chemical shift is also influenced by the density of the electrons around the proton. This electron density is involved in computational chemistry calculations. The 1H-NMR chemical shift calculation between the experiment and the computational method is presented in Table 2. Computational data were generated from the semi-empirical method (AM1, PM3, and PM6), the ab initio method (HF 3-21G, HF 6-31G, and HF 6-311G), and the DFT method based on the 3-21G, 6-31G and 6-311G functional hybrid B3LYP base set. The results showed that the best R2 and PRESS values were obtained using the HF 6-31G method (R2 = 0.98 and PRESS = 0.58). This result clearly indicated that 1H-NMR chemical shift data obtained from a calculation using HF 6-31G has a better agreement with those resulting from experimental measurement than those calculated by other methods. This suggests that the HF 6-31G method presented chalcones' chemical conformation more accurately than the other methods. Therefore, HF 6-31G method has been selected as a calculation method for all breast anticancer compounds in Table 1.
Electronic and molecular descriptor calculations
Electronic and molecular descriptor calculations were carried out by optimizing the molecular geometry of 48 chalcone derivatives in Table 1. Geometry optimization was performed using Gaussian® 09W software with the HF 6-31G methods. There were 18 electronic descriptors obtained, namely 16 net atomic charges, HOMO energy, and LUMO energy. Molecular descriptor calculations were performed using Hyperchem 8.0 software, which produced 6 descriptors, namely molecular surface area, molecular volume, logarithms of partition coefficient (log P), molar refractivity, polarizability, and molecular mass.
Model validation
Using the BuildQSAR program, we searched the total pool of D = 26 descriptors and obtained optimal models shown in Table 2 linking the compounds' molecular structure with their activity. BuildQSAR was a free QSAR program that allowed researchers to build and analyze quantitative models through regression analysis. Multiple regressions were performed using BuildQSAR to correlate physicochemical descriptors and cytotoxic activity of chalcone derivatives. The program automatically detects collinearity between descriptors, and only descriptors with non-collinearity are included in the regression equation. The program can also detect outlier data. No statistical analysis was needed to use BuildQSAR because the analysis was already included in the program (De Oliveira and Gaudio, 2000). Compounds 1, 2, 31, and 37 were detected as outlier data, so they were excluded from both the training and test sets.
Internal validation must satisfy statistical parameters, namely R2> 0.6; Fcal/Ftab > 1, and PRESS was as small as possible. The six models were shown in Table 4 satisfy statistical parameters. The six models were tested externally for validity to determine the model's ability to predict anticancer activity in the test set compounds. From the predicted R2 value and PRESS in the test set shown in Table 5, equation model 2 had the largest R2 and the smallest PRESS, so that the best model was model 2. Model 2 on complete equation was shown as follow:
pIC50 = 3.869 + (1.427 x qC1) + (4. 027 x qC10) + (0.856 x qC15) - (35.900 x ELUMO) + (0.208 x Log P)
n = 36; R2 = 0.743; R2Adj = 0.7002; SEE = 0.198; Fcal/Ftab = 6.423; PRESS = 0.159
Based on the model, it can be seen that to have a more active compound (higher pIC50 value), the value of qC1, qC10, qC15, and Log P, should be increased, but on the contrary, it is decreasing the E_LUMO value. This model will be used to design new chalcone derivatives.
Design of new compound
QSAR model 2 was used as a model to design a new chalcones derivatives with breast anticancer activity of MCF7 cells by replacing the R substituent, as shown in Table 6. From the equation of QSAR model 2, it can be seen that the more positive pIC50 value gave better breast anticancer activity. Based on the QSAR model 2, the descriptors that affect the breast anticancer activity were the net atomic charge on C1, C10, C15, E_LUMO, and log P. The substitution of -H atom by -Cl at the para position can increase the electron density at C1, C10, and C15. Replacement of the -Cl substituent in the ortho or para position can decrease LUMO's E value, increasing the compound's anticancer activity.
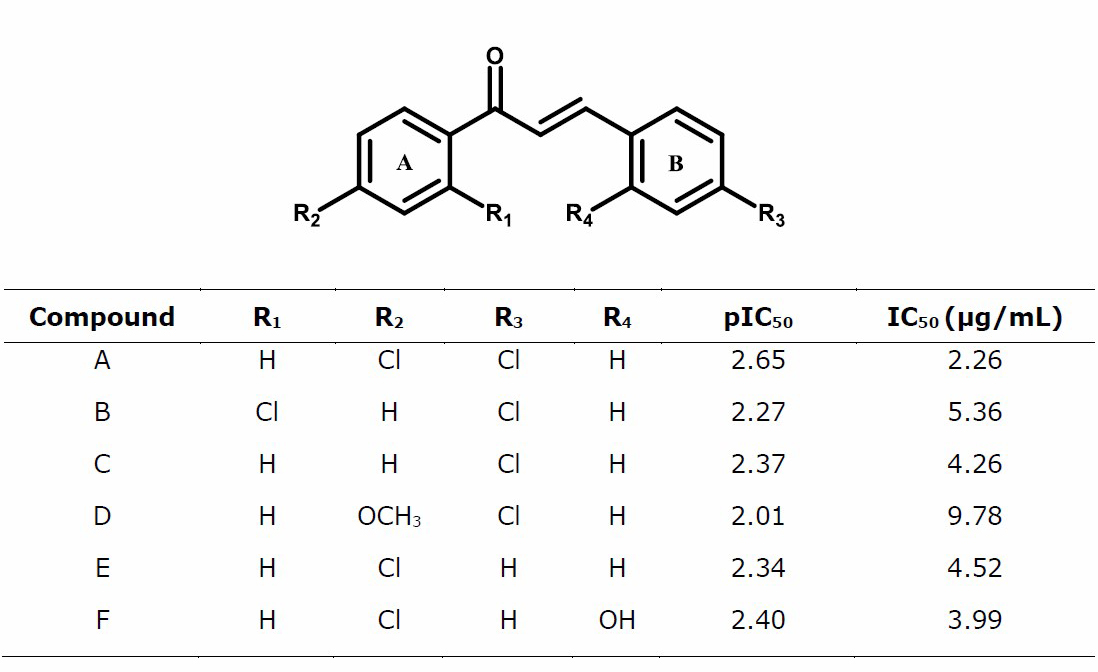
Conversely, the replacement of the electron-donating groups such as -OCH3 and -OH can increase the E LUMO value. Based on experimental studies, the presence of the -Cl substituent at the para position on ring A can increase anticancer activity (Suma et al., 2019). The -Cl substituent in the para position can increase lipophilicity so that the activity increases. Based on experimental studies, modification of chalcone structure using substituent electron-withdrawing groups, namely -F and -Cl, will produce lipophilicity changes that will affect anticancer activity (Ivkovic et al., 2013). Based on the design results in Table 6, the compound that had the best IC50 prediction is chlorochalcone A.
Docking method validation
The validation of the docking protocol was carried out by looking at the simulation's ability to reproduce the ligand co-crystal pose in the target protein. The objective function that is generally used to assess this ability is the RMSD (Root Mean Square Deviation). The parameter used to validate the docking method is RMSD (Marcou and Ragnan, 2007). RMSD depicts the deviation of the docking pose distance compared to the 3D pose of the target ligand calculated and visualized using YASARA. The RMSD value of the docking ligands in Figure 3 showed < 2 Å. It means that the docking parameters were quite accurate and could be used for further processing.
Docking study of newly design chalcones derivatives
Molecular docking simulations were used to determine the interaction of chlorochalcones A-F on the active site of the cyclooxygenase-2 (COX-2) enzyme. The active site of the COX-2 enzyme consists of 3 important regions. First, a hydrophobic pocket consists of amino acid residues Tyr 385, Trp387, Phe 518, Ala201, Tyr248, and Leu352. Second, the region located at the active site gate consists of hydrophilic amino acid residues Arg120, Glu524, and Tyr355, while the third is a site pocket consisted of amino acids His90, Arg513, and Val523 (Krishna et al., 2013). Arg120 and Tyr355 are the important amino acid residue components of the COX-2 enzyme's active site and play a significant role as the ligand's gateway to the enzyme's active site, and then the inhibitory effect is initiated (Kumar et al., 2013). The interaction of native ligands (indomethacin and SC-558) with the COX-2 enzyme's active site occupied these three regions could be seen in Figures 5a and 5b. Indomethacin had an attractive charge interaction with the amino acid residue Arg120 and hydrogen bonds with the amino acid residue Tyr355.
Meanwhile, SC-558 had a hydrogen bond interaction with the amino acid residues Arg120, Gln 192, Leu 352 and a hydrophobic interaction with the amino acid residue Tyr355. Chlorochalcone A-F also occupies the 3 regions the same as the native ligands. There were also interactions with Arg120 and Tyr355; for example, in chlorochalcone A, it can be seen in Figures 6a and 6b. Figure 6a shows that compound A had π cation bond with amino acid residue Arg120 and van der Waals interaction with amino acid residue Tyr355. Figure 6b shows that compound A had a hydrogen bond with Tyr355 and π cation bond with Arg120. In addition, besides Arg120 and Tyr355, chlorochalcone A also interacts with important amino acid residues on the active site of the COX-2 enzyme, namely Tyr385, Ser530, and Trp387. The similarity of amino acid residues indicates that chlorochalcone could occupy the active site of the COX-2 enzyme and is believed to engage in inhibitory activity against the COX-2 enzyme (Miladiyah et al., 2017).
Based on Tabel 7, the interaction energy of chlorochalcones A-F to COX-2 enzymes varies between -87.6766 to-92,2053 for 4COX and -79.4650 to -84.9019 for 6COX with the H-bond found in amino acid residues Arg120, Tyr355, Tyr385, and Met522. This energy is lower than indomethacin (-75.4911) and SC558 (-71.3196). The presence of H-bonds in the amino acid residues Arg120, Tyr355, Tyr385 were also associated with better COX-2 inhibitory activity (Dhingra et al., 2014). As shown in Table 7, it appears that free binding energy throughout chalcones derivatives of COX-2 enzyme is lower than native ligand (Indomethacin and SC-558). It is assumed that interactions of chalcones derivatives with the active site of COX-2 are stronger than native ligand, but of course, this needs further investigation involves experimental laboratory studies.
CONCLUSION
A series of chlorochalcones derivatives have been studied using QSAR and molecular docking simulations to find alternative compounds for breast anticancer drug candidates. A valid QSAR model based on pIC50 against MCF7 cell lines of 48 chalcone derivatives was developed using electronic and molecular descriptors. The best model was used to design new chalcone derivatives that have potential against MCF7 cancer cells. Based on the docking study, the free binding energy in all the new chalcone derivatives of the COX-2 enzyme is lower than the native ligand (Indomethacin and SC-558). It occupies the same active site as the native ligand in the COX-2 enzyme. Interactions of chalcone derivatives with the active COX-2 site are thought to be greater than native ligands. This QSAR and molecular docking recommendation will be proceeded by doing some syntheses and in vitro anticancer assays of the new chlorochalcones.
ACKNOWLEDGEMENT
The Financial support from The Ministry of Research, Technology, and Higher Education of The Republic of Indonesia via Penelitian Disertasi Doktor scheme (PDD, Contract No.: 2280/UN1/DITLIT/DIT-LIT/PT/2021) is gratefully acknowledged. Anita Dwi Puspitasari acknowledged receipt of the Indonesia Endowment Fund for Education (LPDP) Scholarship, Ministry of Finance, Indonesia.
AUTHOR CONTRIBUTIONS
Tutik Dwi Wahyuningsih and Harno Dwi Pranowo conceived the original idea and supervised the project. Anita Dwi Puspitasari planned and carried out the simulations, analyzed the data, and wrote the manuscript with support from Harno Dwi Pranowo, Tutik Dwi Wahyuningsih, and Endang Astuti. All authors discussed the results and contributed to the final version of the manuscript.
CONFLICT OF INTEREST
This research has no conflict of interest with any party.
REFERENCES
Abou-Zied, H.A., Youssif, B.G.M., Mohamed, M.F.A., Hayallah, A.M., and Abdel-Aziz, M. 2019. EGFR inhibitors and apoptotic inducers: design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid Moleculs. Biorganic Chemistry. 89: 1-11.
Das, M., and Manna, K. 2016. Chalcone scaffold in anticancer armamentarium: a molecular insight. Journal of Toxicology. 2016: 1–14.
De Oliveira, D.B., and Gaudio, A.C. 2000. BuildQSAR: A New Computer Program for QSAR Analysis. Quantitative Structure-Activity Relationships. 19: 599–601.
Dimmock, J.R., Ellias, D.W., Beazely, M.A., and Kandepu, N.M. 2000. Bioactivities of chalcones. Current Medicinal Chemistry. 6: 1125–1149.
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., et al. 2016. Gaussian 09. Revision A.02. Gaussian, Inc., Wallingford CT.
Globocan. 2018. Estimated cancer incidence, mortality, prevalence and disability-adjusted life years (DALYs) worldwide in 2018. IARC Cancer Base No. 11.
Gurrapu, N., Kumar, E.P., Kolluri, P.K., Putta, S., Sivan, S.K., and Subhashini, N.J.P. 2020. Synthesis, biological evaluation and molecular docking studies of novel 1,2,3-triazole tethered chalcone hybrids as potential anticancer agents. Journal of Molecular Structure. 1217: 1-12.
Ivkovic, B.M., Nikolic, K., Ilic, B.b., Zizak, Z.S., Novakovic, R.b., Cudina, O.A., and Vladimirov, S.M. 2013. Phenylpropiophenone derivatives as potential anticancer agents: synthesis, biological evaluation and quantitative structure activity relationship study. European Journal of Medicinal Chemistry. 63: 239–255.
Jandial, D.D., Blair, C.A., Zhang, S., Krill, L.S., Zhang, Y.B., and Zi, X. 2014. Molecular targeted approaches to cancer therapy and prevention using chalcones. Current Cancer Drug Targets. 14: 181–200.
Khanapure, S., Jagadale, M., Bansode, P., Choudari, P., and Rashinkar, G. 2018. Anticancer activity of ruthenocenyl chalcones and their molecular docking studies. Journal of Molecular Structure. 142-147.
Khrishnamachary, B., Stasinopoulus, I., Kakkad, S., Penet, M.F., Jacob, D., Wildes, F., Mironchik, Y., Pathak, A.P., Solaiyappan, M., and Bhujwalla, Z.M. 2017. Breast cancer cell cyclooxygenase-2 expression alters extracellular matrix structure and function and numbers of cancer associated fibroblasts. Oncotarget. 8: 17981–17994.
Krishna, P.S., Vani, K., Prasad, M.R., Samatha, B., Bindu, N.S.V.S.S.S.L.H., Charya, M.A.S.C., and Shetty, P.R. 2013. In-silico molecular docking analysis of prodigiosin and cycloprodigiosin as COX-2 inhibitors. SpringerPlus. 2: 172–177.
Kumar, V., Gupta, G.K., Kaur, K., and Singh, R. 2013. 4-Fluorophenylhydrazones as potential COX-2 inhibitors: a novel, efficient, one pot solid phase synthesis, docking study and pharmacological evaluation. Medicinal Chemistry Research. 22: 5890–5900.
Korb, O., Stutzle, T., and Exner, T.E. 2009. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. Journal of Chemical Information and Modeling. 49: 84–96.
Marcou, G. and Ragnan, D. 2007. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. Journal of Chemical Information and Modelling. 47: 195–207.
Marquina, S., Santiago, M.M., Caranza, J.N.S., Mojica, M.A., Maya, L.G., Hernandez, R.S.R., and Alvarez, L. 2019. Design, synthesis and QSAR study of 2′-hydroxy-4′-alkoxy chalcone derivatives that exert cytotoxic activity by the mitochondrial apoptotic pathway. Bioorganic & Medicinal Chemistry. 27: 43–54.
Miladiyah, I., Jumina, J., Haryana, S.M., and Mustofa, M. 2017. In silico molecular docking of xanthone derivatives as cyclooxygenase-2 inhibitor agents. International Journal of Pharmacy and Pharmaceutical Sciences. 9: 43–54.
Onlu, S., and Sacan, M.T. 2017. Impact of Geometry Optimization Methods on QSAR Modelling: A case Study for Predicting Human Serum Albumin Binding Affinity. SAR and QSAR in Environmental Research. 28: 491–509.
Pingaew, R., Saekee, A., Mandi, P., Nantasenamat, C., Prachayasittikul, S., Ruchirawat, S., and Prachasittikul, V. 2014. Synthesis biological evaluation and molecular docking of novel chalcone-coumarin hybrids as anticancer and antimalarial agents. European Journal of Medicinal Chemistry. 85: 65-76.
Raghavan, S., Manogaran, P., Kuppuswami, B.K.M., Venkatraman, G., and Nurasimha, K.K.G. 2015. Synthesis and anticancer activity of chalcones derived from vanilin and isovanillin. Medicinal Chemistry Research. 24: 4157–4165.
Rasyid, H., Purwono, B., and Armunanto, R. 2018. Quantitative structure activity relationship (QSAR) based on electronic descriptors and docking studies of quinazoline derivatives for anticancer activity. Oriental Journal of Chemistry. 34: 2361–2369.
Rybka, M., Mercader, A.G., and Castro, E.A. 2014. Predictive QSAR study of chalcone derivatives cytotoxicity activity against HT-29 human colon adenocarcinoma cell lines. Chemometrics and Intelligent Laboratory Systems. 132: 18–29.
Shenvi, S., Kumar, K., Hatti, K.S., Rijesh, K., Diwakar, L., and Reddy, G.C. 2013. Synthesis, anticancer and antioxidant activities of 2, 4, 5 – trimethoxy chalcone and analogues from asaronaldehyde: structure-activity. European Journal of Medicinal Chemistry. 62: 435–442.
Sokalingam, S., Munussami, G., Kimi, J.R., and Lee, S.G. 2018. Validation on the molecular docking efficiency of lipocalin family of protein. Journal of Industrial and Engineering Chemistry. 67: 293–300.
Soslow, R.A., Dannenberg, A.J., Rush, D., Woerner, B.M., Khan, K.N., Masferrer, J., and Koki, A.T. 2000. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 89: 2637–2645.
Suma, A.A.T., Wahyuningsih, T.D., and Mustofa. 2019. Efficient synthesis of chloro chalcones under ultrasound irradiation, their anticancer activities and molecular docking studies. Rasayan Journal of Chemistry. 12: 502–510.
Syam, S., Abdelwahab, S.I., Al-Mamary, M.A., and Mohan, S. 2012. Synthesis of chalcones with anticancer activities. Molecules. 17: 6179–6195.
Veerasamy, R., Rajak, H., Jain, A., Sivadasan, S., Varghese, C.P., and Agrawal, R.K. 2011. Validation of QSAR models – strategies and importance. International Journal of Drug Design and Discovery. 2: 511–519.
Yadav, D.K., Ahmad, I., Shukla, A., Khan, F., Negi, A.S., and Gupta, A. 2013. QSAR and docking studies on chalcone derivatives for antitubercular activity against M. Tuberculosis H37Rv. Journal of Chemometrics. 28: 499–507.
Wild, C.P., Weiderpass, E., and Stewart, B.W. 2020. World cancer report: cancer research for cancer prevention. Lyon, France: International Agency for Research on Cancer. Available from: http://publications.iarc.fr/586. Licence: CC BY-NC-ND 3.0 IGO.
OPEN access freely available online
Chiang Mai University Journal of Natural Sciences [ISSN 16851994]
Chiang Mai University, Thailand
https://cmuj.cmu.ac.th
Anita Dwi Puspitasari1,2,3 , Harno Dwi Pranowo1,2, Endang Astuti1 and Tutik Dwi Wahyuningsih1,*
1 Department Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
2 Austria-Indonesia Centre for Computational Chemistry, Universitas Gadjah Mada, Sekip Utara, Yogyakarta 55281, Indonesia
3 Faculty of Pharmacy, Universitas Wahid Hasyim, Semarang, 50236, Indonesia
Corresponding author: Tutik Dwi Wahyuningsih, E-mail: tutikdw@ugm.ac.id
Total Article Views
Editor: Korakot Nganvongpanit,
Chiang Mai University, Thailand
Article history:
Received: February 4, 2021;
Revised: April 14, 2021;
Accepted: April 22, 2021;
Published online: May 11 , 2021