 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Insight on the In Silico Study and Biological Activity Assay of Chalcone-Based 1, 5-Benzothiazepines as Potential Inhibitor for Breast Cancer MCF7
Neni Frimayanti*, Marzieh Yaeghoobi, Ihsan Ikhtiarudin, Dhea Rizki Wannisyah Putri, Hamid Namavar, and Fatemeh S.BitarafPublished Date : 2020-12-17
DOI : https://doi.org/10.12982/CMUJNS.2021.019
Journal Issues : Number 1, January-March 2021
Abstract In silico study was performed to twelve 1,5-benzothiazepine chalcone derivatives with the protein target from the crystallographic structure modeling of the enzyme tyrosine kinase. The objective of this study is to execute and to estimate the biological activity of chalcone-based 1,5-benzothiazepine derivatives as potential inhibitors for breast cancer MCF7. To get insight into potential anticancer activities, molecular docking, molecular dynamic and ADME prediction were performed. Docking results reported that compound MA9 with binding free energy of -11.2 kcal / mol can interact through hydrogen bonds with amino acids Cys788 on 1T46 protein active site. In addition, the lowest binding free energy conformation indicated its stability during molecular dynamic simulation. MA9 is also shown to have drug likeness properties based on ADME prediction. In order to evaluate the modeling outcomes, MTT assay were performed for some of the most and least promising benzologs (i.e., MA1, MA6, MA8 and MA9). As expected, compound MA9 with the best calculated anticancer properties revealed the best inhibition against MCF7cell line in vitro. Thus, this compound was chosen as the reference for the next stage in the drug design.
Keywords: ADME, Benzothiazepine, Docking, MCF7, Molecular Dynamic
Citation: Frimayanti, N., Yaeghoobi, M., Ikhtiarudin, I., Putri, D.R.W., Namavar, H., and Bitaraf, F.S. 2021. Insight on the in silico study and biological activity assay of chalcone-based 1, 5-benzothiazepines as potential inhibitor for breast cancer MCF7. CMUJ. Nat. Sci. 20(1): e2021019.
INTRODUCTION
Cancer is one of the leading causes of death worldwide. It is an abnormal growth of body tissue cells that turn into malignant cells that can grow further and spread to other parts of the body and may result death. Breast cancer is one type of cancer; which is very frightening for women throughout the world. According to The American Cancer Society, in the incidence of invasive breast cancer in 2018 occurred as many as 266.120 cases in women, 2.550 cases in men, 63.960 cases of carsinoma in situ (CIS) and 40.610 cases (14%) deaths due to breast cancer (Rebbeca et al., 2018).
Benzothiazepines are seven membered heterocyclic compounds, containing nitrogen and sulfur (Vyawahare et al., 2010) and 1, 5-benzotiazepine derivatives are important pharmacophores from this family (Kurokawa et al., 1997). According to IUPAC (International Union of Pure and Applied Chemistry), pharmacophore is a steric and electronic factor needed to ensure the optimal molecular interactions with specific biological target structures (Mannhold et al., 2006; Narendran et. al., 2014). 1, 5-Benzothiazepine is a pharmacophore that can provide a broad spectrum of biological activity such as Ca2+ channel antagonists (Kurokawa et al., 1997), antimicrobials (Gaikwad et al., 2013), lung cancer (Ameta et al., 2013) and anticonvulsants (Garg et al., 2010). Rationally, in silico studies are playing important role in drug design (Neni et al., 2020). In the process of drug design, in silico studies (i.e. molecular docking and molecular dynamic) are used to find the best conformation with low binding free energy. In addition, they can provide useful information for predicting the orientation of a candidate drugs bond to its target (i.e. protein). To date, although some in silico investigations have been carried out on potential anticancer properties of chalcone derivatives like pyrazolines (Jasril et al., 2017) and quercetins (Lelita et al., 2017) but unfortunately, thus far, chalcone-derived heterocycles like 1,5-benzothiazepines have not been extensively studied for their potential anticancer activities. Moreover, there are not many reports on discovering an anticancer drugs using in silico tools such as molecular docking, molecular dynamic (MD) and ADME calculation. This study aimed to explore new potential agents from 1, 5-benzothiazepine against breast cancer MCF7 using tyrosine kinase as the target.
MATERIALS AND METHODS
Molecular docking
All 1, 5-benzothizepines were synthesized, purified and characterized following reported procedure (Yaeghoobi, 2012). The molecular structure of these benzologs and Doxorubicin as positive control were drawn using Chemdraw Ultra 13.0 and stored in "cdx" format and converted to PDB format using Discovery studio visualizer (DSV) software package. The energies of all the ligands were minimized for then the minimized structures were subsequently prepared with detected root of torsion and number of torsion. Molecular structure of twelve 1, 5-benzothiazepine and doxorubicin are shown in Table 1.
Table 1. Molecular structures of 1, 5-benzothiazepine
The structure of protein was downloaded from the protein databank (www.pdb.org PDB ID: 1T46), then the water molecules and initial ligands in the protein were removed. Further preparation carried out by adding hydrogen atoms, polar only and merged non polar. A grid box of the protein structure was then achieved with a grid spacing of 1 Å, dimensions of 31 x 25 x 35 points along the x, y and z-axes and centered on the protein active site. Docking was performed using Autodock vina software packages (Trott et al., 2010).
Molecular dynamic simulation
NAMD (NAnoscale Molecular Dynamics program v 2.9) was excreted to execute the molecular dynamic simulation (i.e. preliminary study). The selected force field of CHARMM27 (Chemistry at HARvard Macromolecular Mechanics) was utilized as the best force field. TIP3P water box with 2.5 Å water layer for each direction of coordinated structure was used to achieve modeled protein.
NVT ensemble from 0 to 300 K over 100 ps was applied to heat the system gradually. MD simulations was conducted on 50 ns time scale for each system in an isothermal isobaric ensemble (NPT) with periodic boundary conditions. The coupling of temperature and pressure parameters were set on 1.0 ps. The coordinates were saved at every 0.1 ps during the sampling process. The simulations were generated the conformations for then they were used for furhter binding free energy calculations and decomposition process.
Adsorption, distribution, metabolism and excretion (ADME) prediction
ADME profiles of potential compounds were calculated to get better insight into the physicochemical and pharmacokinetic properties as well as prediction of drug likeness of candidates. In this study, the ADME profiles were calculated using SwissADME server (http://www.swissadme.ch/index.php).
Biological assay using MTT assay
To evaluate the modeling results, MTT assay were performed for compounds with the most and least MCF7 cells were seeded into 96-well plates at an initial density of 104 cells/well. MA1, MA6, MA8, MA9 and Doxorubicin were then added as standard drug to achieve a final concentration of 100, 50, 25, 10, 5, 2.5, or 1.25 μgr/ml. MTT( Methyl Thiazole Tetrazolium) was dissolved in PBS to obtain the concentration of 5 mg/mL, then it filter sterilized and added in a volume of 10 μL to each well to attain the final concentration of 0.5 mg/mL and incubated for 4 h. The MTT-containing medium was replaced by 100 μL of dimethylsulfoxide (DMSO, SantaCruz, Dallas, TX, USA), and the plates were gently agitated for 10 min. Absorbance was obtained at 570 nm with background subtraction at 690 nm using Biotek Cytation 5imaging reader (Biotek Instruments, Winooski, VT, USA). All of the experiments were performed in triplicate.
RESULTS
Molecular docking
Molecular docking is a good tool to predict and match the desired binding sites, understanding possible conformation of the compounds and further to clarify the binding interactions between ligand and receptor.
In this study, twelve 1,5-benzothiazepines were docked as ligands into protein (PDB ID: 1t46) to elucidate the ligand interactions with protein binding site. The docking results are listed in Table 2 which highlighted the tendency of these chalcone-based benzologs to bind tightly to the active site as well as allosteric sites of tyrosine kinase as receptor. The best poses of complex ligand-protein were selected according to the lowest value of binding free energy and also Root Mean Squared Deviation (RMSD) value. Based on the docking results, MA9 estimated to be active as breast cancer MCF7 inhibitor. Although MA5 has also displayed a hydrogen bond with Cys788 residue, yet, its binding free energy value is higher than the positive control and it cannot be considered as a potentially active ligand. In addition, compounds MA1 and MA6 have shown the binding free energy value of -9.3kcal/mol and -9.1kcal/mol, respectively. However, MA1 has not exhibited any hydrogen bonding or other interaction with amino acid residue. Although MA6 has displayed hydrogen bonds with Asp810 and Glu 640, nevertheless, these bindings are different from the amino acid sequence of the positive control. Hence, these compounds can not be considered as potential inhibitors for MCF7.
Table 2. The docking results.
|
Compound |
Parameters |
||
|
Binding free energy (kcal/mol) |
RMSD value |
Hydrogen bond |
|
|
MA1 |
-9.3 |
0.000 |
- |
|
MA2 |
-9.4 |
0.000 |
- |
|
MA3 |
-10.2 |
0.000 |
- |
|
MA4 |
-9.9 |
0.000 |
- |
|
MA5 |
-9.3 |
0.000 |
Cys788 |
|
MA6 |
-9.1 |
0.000 |
Asp810, Glu640 |
|
MA7 |
-9.7 |
0.000 |
- |
|
MA8 |
-10.1 |
0.000 |
Cys788 |
|
MA9 |
-11.2 |
0.000 |
Cys788 |
|
MA10 |
-10.0 |
0.000 |
- |
|
MA11 |
-9.8 |
0.000 |
- |
|
MA12 |
-10.0 |
0.000 |
- |
|
Doxorubicin |
-9.9 |
0.000 |
Asp677, Tyr672 |
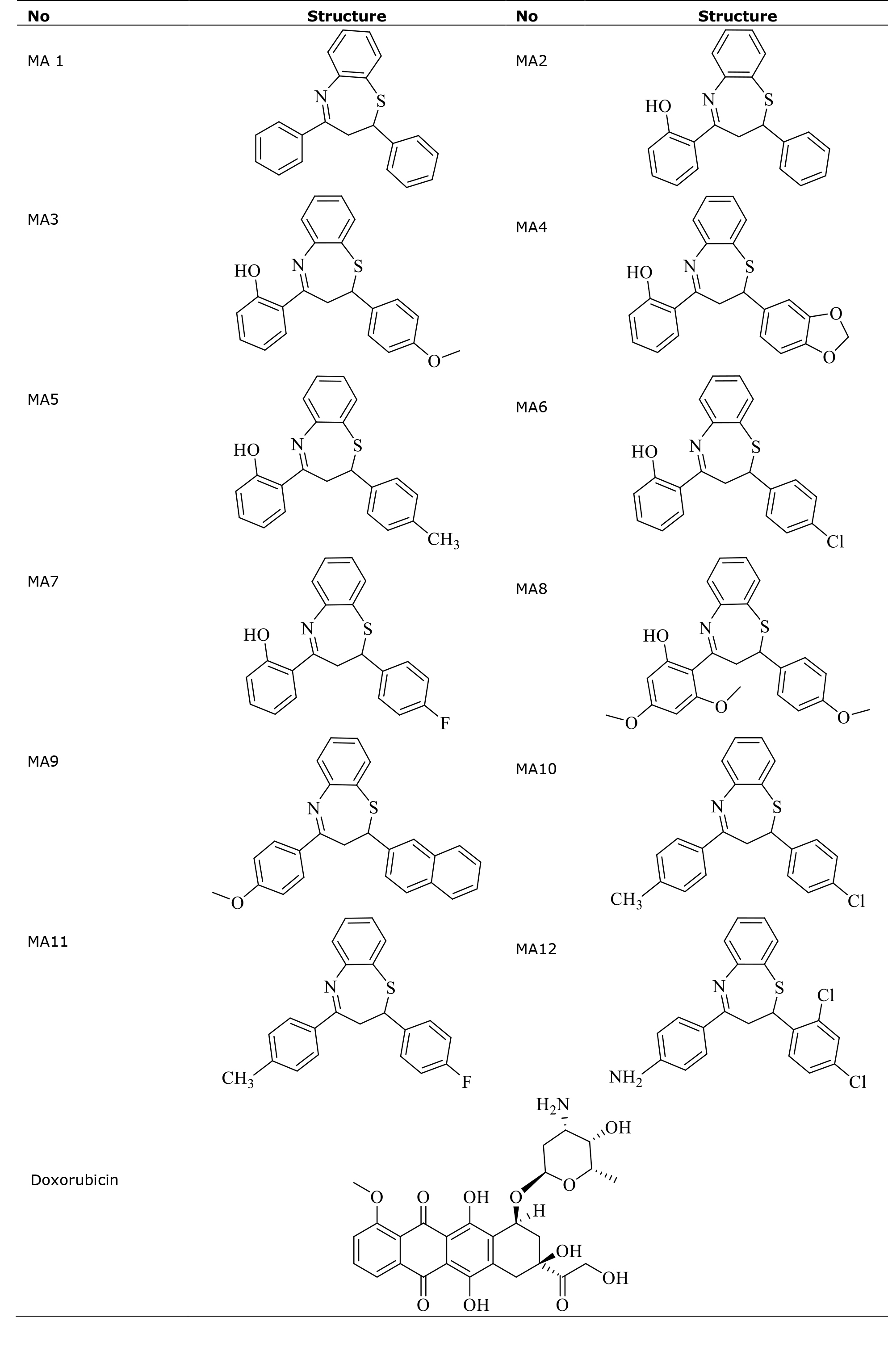
Compound MA8 with binding free energy value of -10.3kcal/mol, was observed to have one hydrogen bonding with amino acid residue Cys788, van der Waals interaction with Ser639 Glu640, Leu644, Tyr570, Ile571 and Ile789. The spatial arrangement of MA8 is presented in Figure 1.
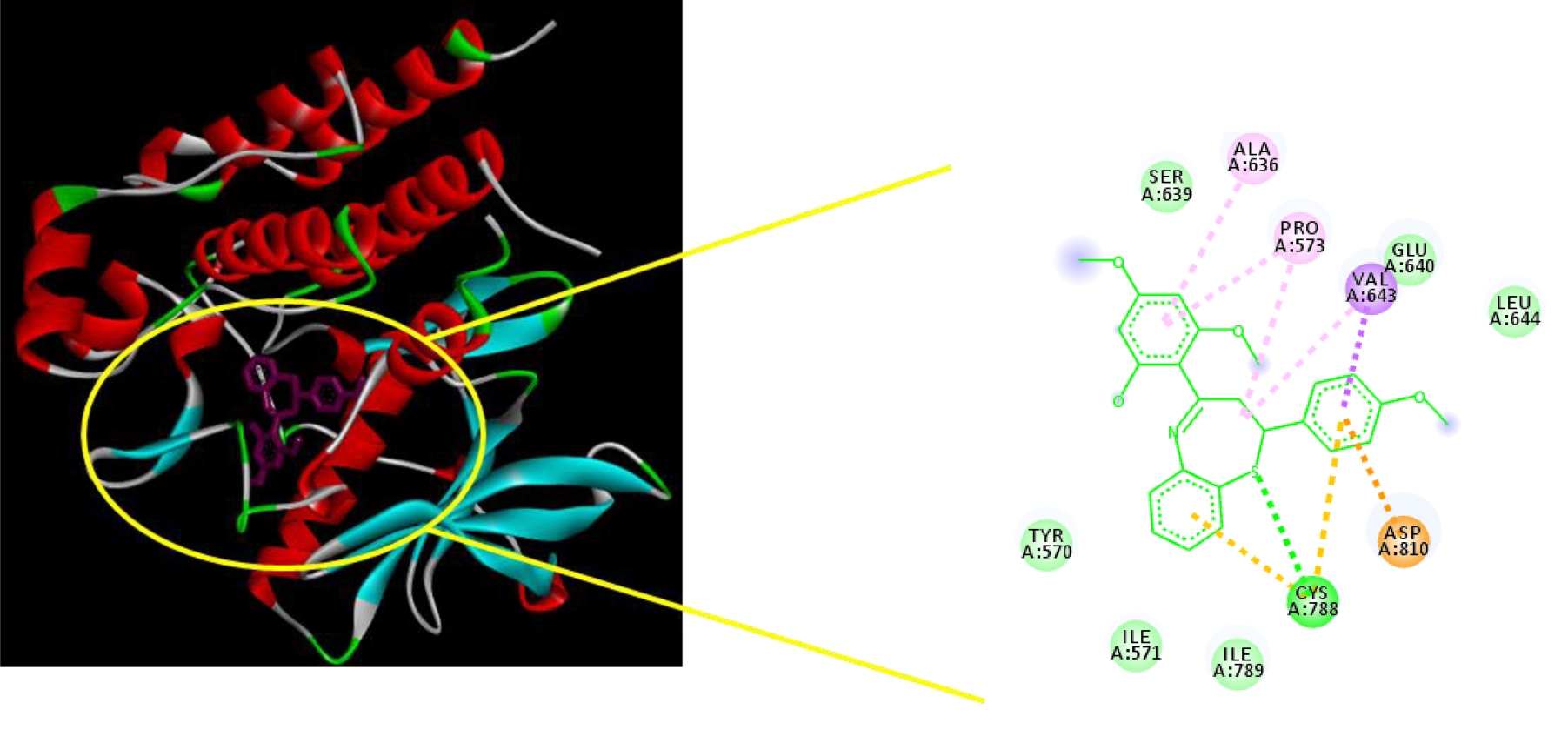
Figure 1. Spatial arrangement of compound MA8
The estimated active compound, MA9, with binding free energy value of -11.4kcal/mol, interacted via hydrogen bonding with Cys788. Furthermore, MA9 is exhibited to interact through van der Waals interaction with amino acid Glu640, Ser639, Leu647, Leu783, Ile571 dan Tyr570. Thus, MA8 and MA9 predicted to be active compounds. The existence of methoxy groups in ring A may enhance the hydrophobicity of these molecules, which may presumably cause compounds MA8 and MA9 to have better binding affinity compare to other compounds (Noval et al., 2020). Figure 2 demonstrated the spatial arrangement for MA9 which helps to understand the binding site of this ligand with the protein active site.
Superimpose was performed to check the orientation of MA8 and MA9 ligands simultaneously. It seems that both of them have the same orientation to bind with the protein where in both cases, the seven-membered ring flipped slightly. However, other parts of chalcone backbones (i.e. rings A and B) maintained their orientations. Superimpose for MA8 and MA9 shown in Figure 3.
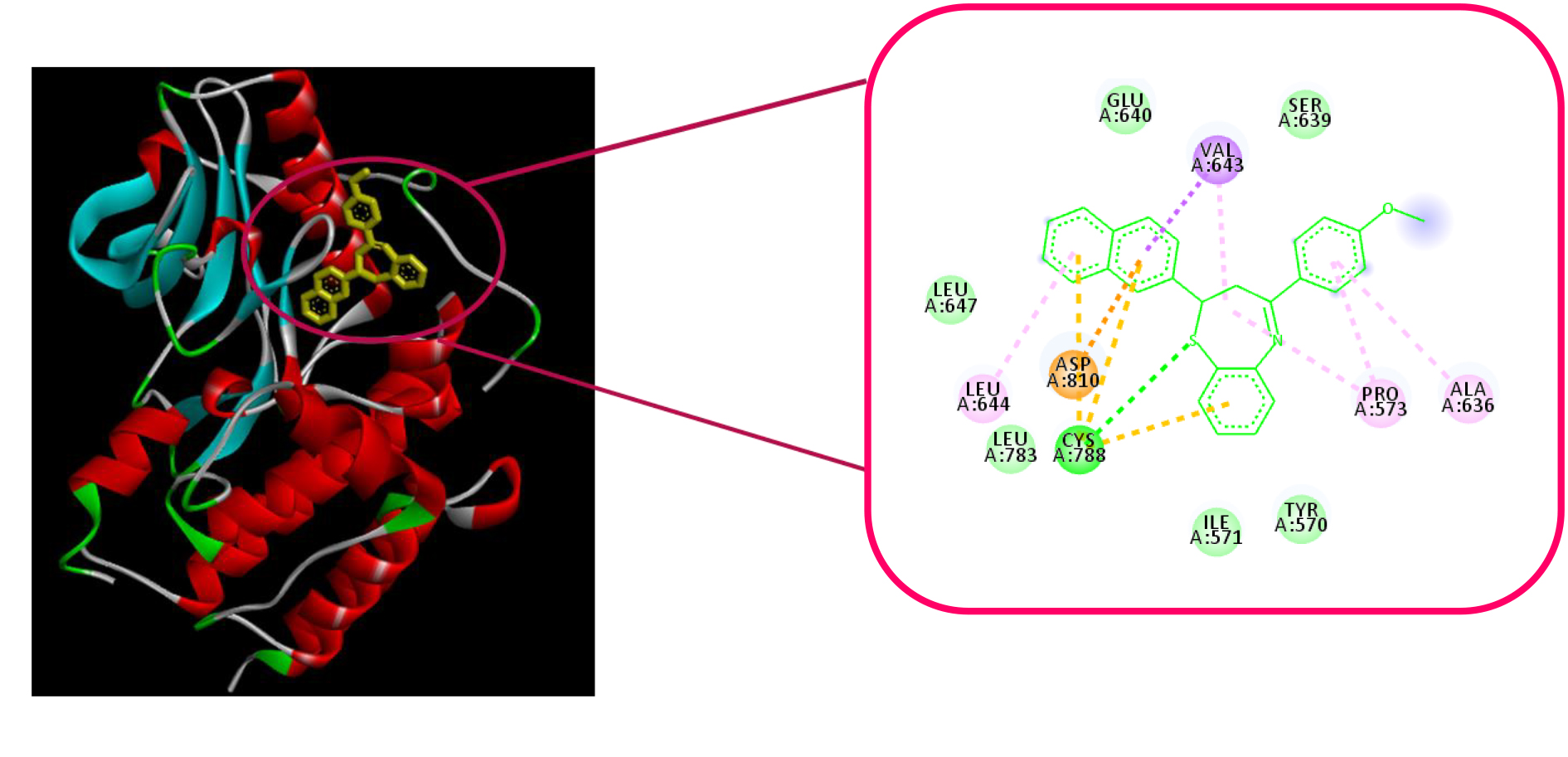
Figure 2. Spatial arrangement of compound MA9.
Figure 3. Superimpose for MA8 (pink) and MA9 (purple).
Based on the docking results, it was found that doxorubicin has binding free energy value of -9.9kcal/mol and an RMSD value of 0,000. Docking visualization results shown that Doxorubicin (i.e. positive control) can bind to 13 amino acid residues on the active site of the receptor, namely Glu671, Ala621, Leu799, Val603, Leu595, Phe811, Asp677, Asn680, Cys674, Tyr657, Gly676, Cys673 and Cys788. It interacts with receptors through hydrogen bonds with amino acid residues Asp677 and Tyr672, van der Waals interaction with residues Cys673, Gly676, Tyr675, Asn689, Cys788 and Phe811, pi-alkyl bonds with residues Ala621, Leu799 Leu595 and Val603, pi-sigma bonds with Leu 799 and Leu595 residues, and pi-donor hydrogen bonds with Asp677 residues. Figure 4 is depicted the spatial arrangement for Doxorubicin.
According to the result, the binding free energies for MA8 and MA9 are lower than Doxorubicin and moreover, they both exhibited crucial binding affinities (i.e. hydrogen bond) with residue Cys788 at the protein active site which seems to be significant for activity prediction. Hence, MA8 and MA9 considered potentially active as MCF7 inhibitors and can be further utilized in drug designing process.
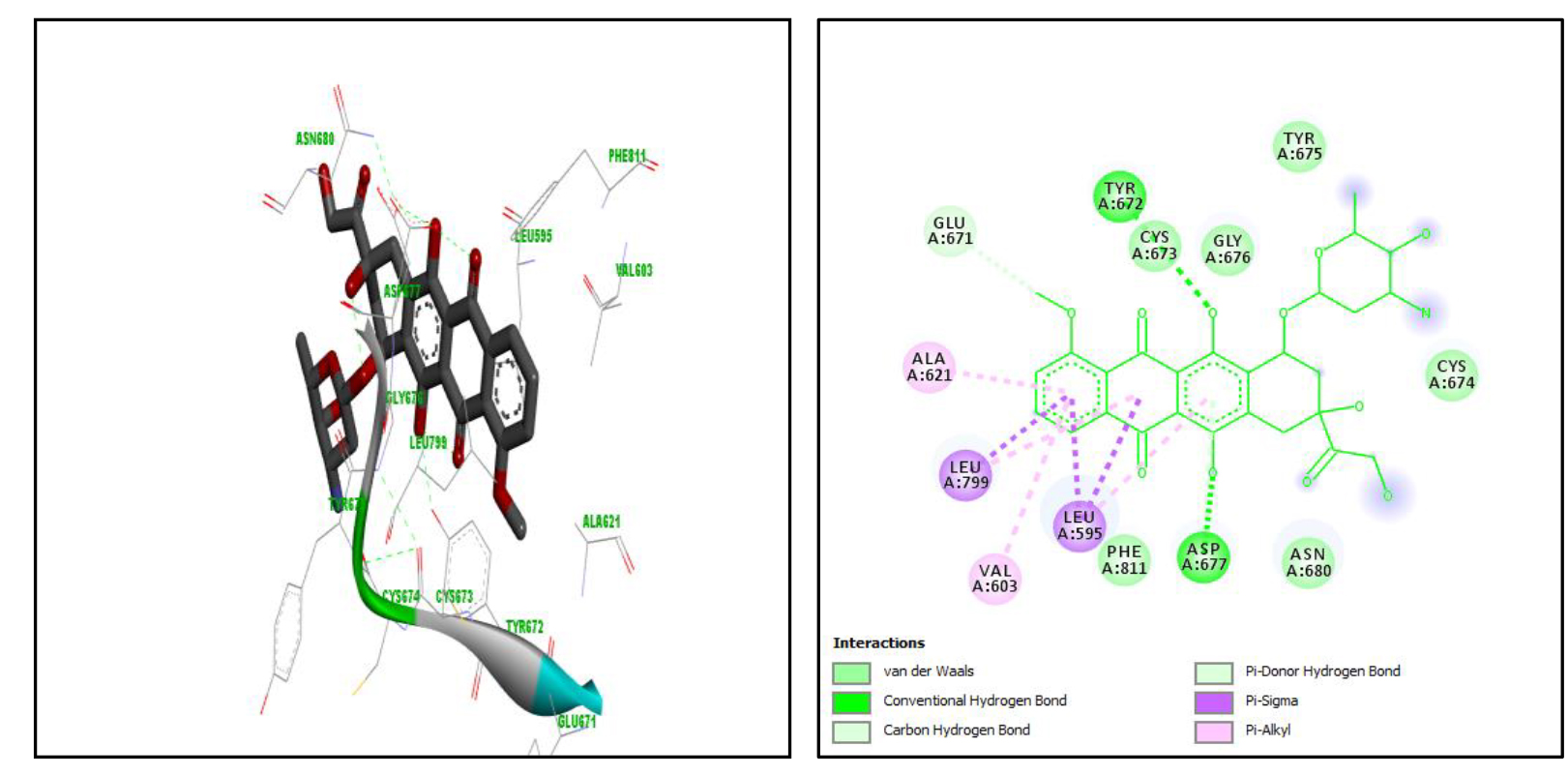
Figure 4. Spatial arrangement of doxorubicin.
Molecular Dynamic
MD simulation was inspected on these two compounds to explore the interactions between ligand and receptor (Adel, et. al 2019). The stability of MD simulation was examined to confirm the binding profile of ligands and to give an overall impression about the estimated active 1,5-benzothiazepine, MD simulation was subjected to run for 50 ns. In the present study, MD simulation was performed to ensure that interaction between protein and active compounds are still maintained (Neni et al., 2020). It was initiated using the high stability with energy minimum at temperature of 300K to see affinity of the ligand to binding site.
The results were inspected by examining the efficiency of hydrogen binding in both potentially active compounds i.e. MA8 and MA9 and also the most inactive compounds i.e. MA1 and MA6 before and after 50 ns and 300K MD simulation. Generally, in case of compound MA9, the conformation was maintained to bind well with the same residues before and after operation and the hydrogen bond distance is less than 2.9 Å. However, compound MA8 has hydrogen bond distance higher than 2.9 Å and the rest of tested benzologs seemed to lose their activity due to the presence of some interactions between ligands and receptor that were not maintained. According to the docking results, MA8 has potency as breast cancer inhibitor, unfortunaely according to MD simulation this compound was not able to maintain the existence of hydrogen bond and the distance of hydrogen bonding is higher than 2.9 Å (Duan et. al., 2019). Table 3 is listed the interactions with amino acid after the MD simulation. Based on MD simulation, MA9 seemed to retain its interactions with the same amino acids after simulation which indicates that it can be used as potential inhibitors for breast cancer. Visualization of MD simulation is depicted in Figure 5.
Table 3. Ligand interaction with amino acid residues.
|
Compound |
After docking |
MD simulation |
Distance of Hbond |
|
MA1 |
- |
- |
- |
|
MA6 |
Glu 640, Asp810 |
- |
- |
|
MA8 |
Cys788, Ser639, Glu640, Leu644, Tyr570, Ile571, Ile789, Asp810, Val643, Pro573, Ala636. |
Ala636, Pro573, Tyr570, Cys788, Asp810, Ile571, Glu640, Leu644, |
3.2 Å |
|
MA9 |
Cys788, Glu640, Ser639, Leu647, Leu783, Ile571, Tyr570, Asp810, Val643, Cys788, Ala636, Pro573 Leu644 |
Ala636, Pro573, Tyr570, Cys788, Asp810, Ile571, Glu640, Leu644, Ser639 |
2.9 Å |
ADME profiles calculation
The in silico absorption, distribution, metabolism and excretion (ADME) profile is also called as pharmacokinetic profile. Hence, ADME was used to expect reduce the risk of late-stage attrition of drug development and to optimize the screening process. However, it only can be used for the promising compounds (Yamashita et al., 2004).
A compound can be determined to have bioavailability if it follows the Lipinski’s rule of five. This rule consisted when the maximum MW is 500, Log P is not greater than 5, hydrogen bond donor is less than 5 and hydrogen bond acceptor is less than 10 (Lipinski et al., 2012).Lipinski’s rule of five calculation was performed to achieve the absorption levels of potentially active compounds to across the lipid bilayers in the human body. ADME profile was generated the drug likeness properties based on Lipinski’s rule of five for compounds MA1, MA6, MA8 and MA9 (Table 4). Based on calculations, the potentially active compounds shown the reasonable drug properties. It is indicated that MA9 is safe to be used as potential anti cancers for MCF7.
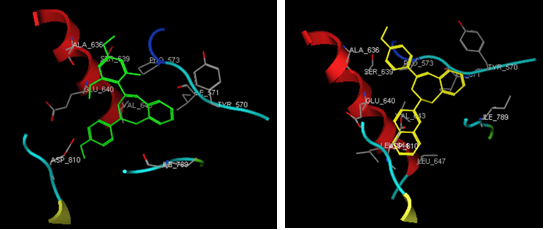
Figure 5. MD Visualization for (a) MA8 and (b) MA9
Table 4. Drug likeness properties of MA8 and MA9.
|
Profiles |
Compound MA1 |
Compound MA6 |
Compound MA8 |
Compound MA9 |
|
MW (g/mol) |
330.47 |
380.91 |
421.51 |
395.52 |
|
Consensus Log Po/w |
3.79 |
3.91 |
3.44 |
3.95 |
|
Hydrogen bond donor |
1 |
2 |
1 |
0 |
|
Hydrogen bond acceptor |
0 |
1 |
5 |
2 |
|
Rotatable bonds |
2 |
2 |
5 |
3 |
|
Drug likeness (Lipinski) |
Yes |
Yes |
Yes |
Yes |
Bological assay
The effect of MA1, MA6, MA8, MA9 and Doxorubicin at various concentrations (100, 50, 25, 10, 5, 2.5, or 1.25 μgr/ml) on the MCF7 cell viability were determined using MTT assay. MCF7 cells incubated for 72 h. The IC50 value for MA9, MA8, MA1 and MA6 were obtained at the concentrations about 550 and 100μgr/ml respectively, whereas the IC50 value for Doxorubicin was observed at about 10μgr/ml. Furthermore, at 5μgr/ml, the cell survival assessed value for MA9 was significantly lower than the rest of tested compounds. The result revealed that amongst all tested compounds, MA1, MA6 and MA8 were confirmed to be inactive while MA9 with the lowest concentration for IC50, exhibited the higher chance of cell survival. Hence, MA9 with the best experimental result can be considered as a potential lead for breast cancer treatment. The MTT assay results summarized in Figure 6 (a and b)
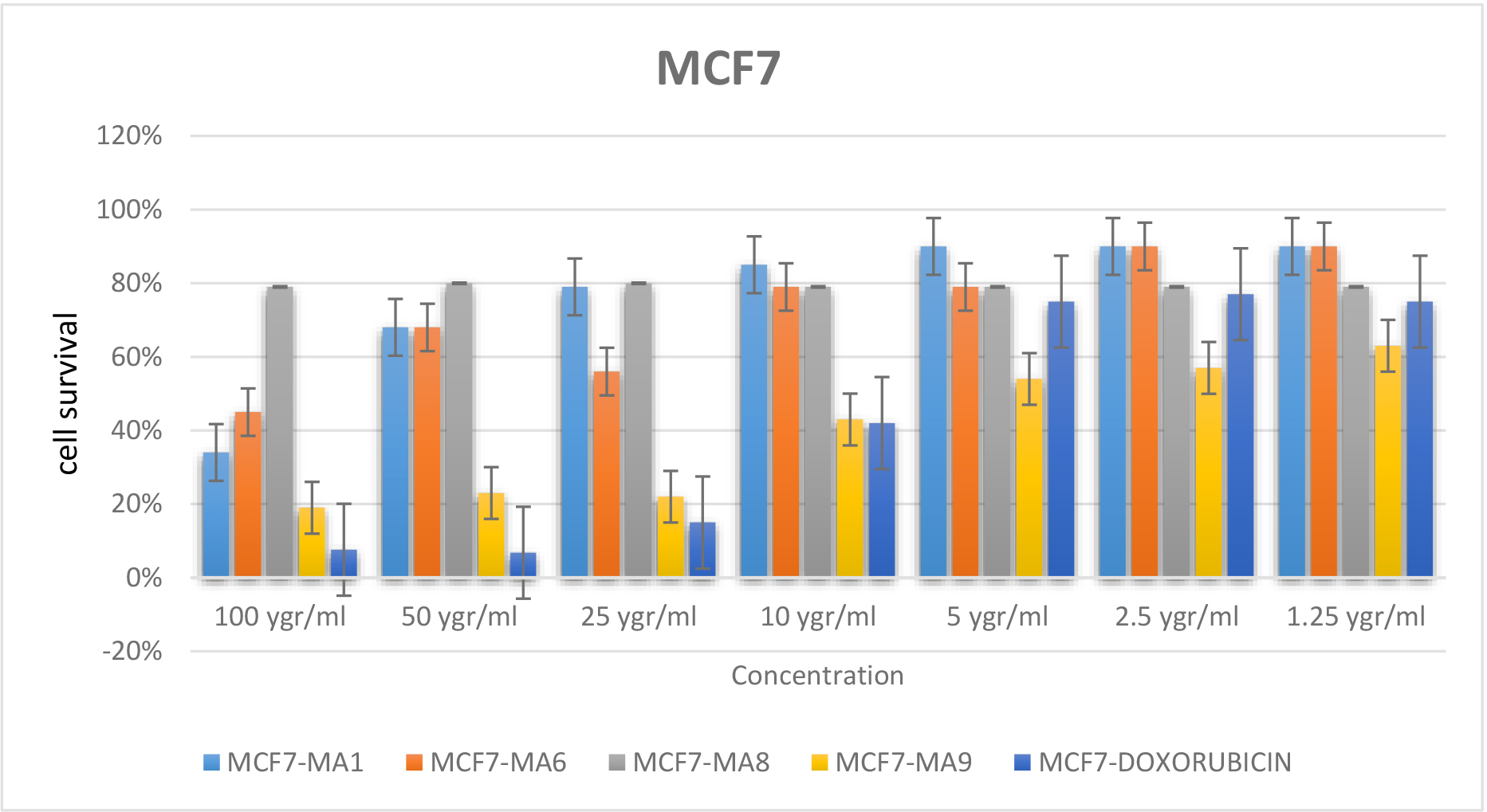
(a)
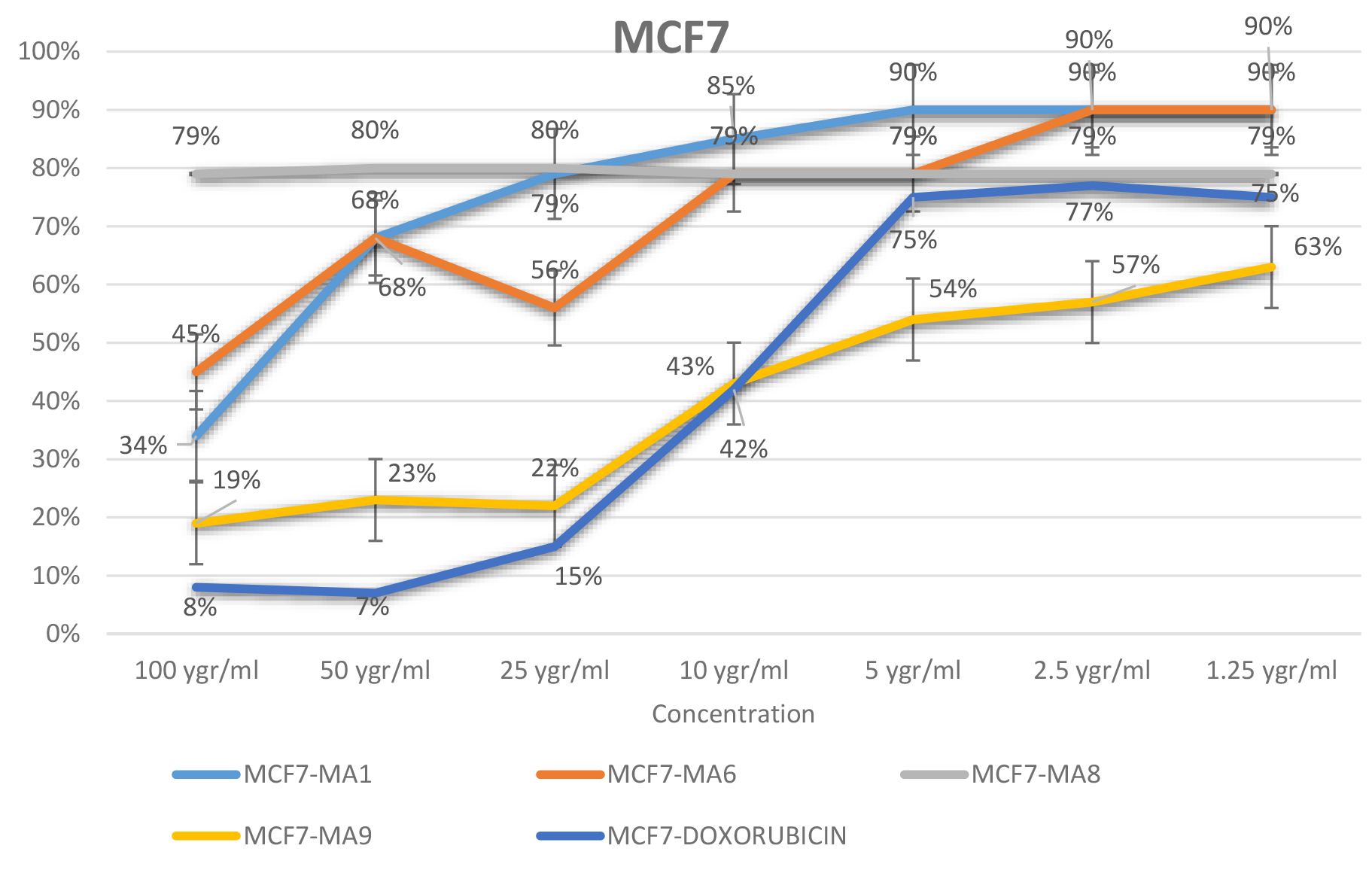
(b)
Figure 6. Percentage of cell survival for tested compounds and Doxorubicin in various concentrations (a) and (b)
CONCLUSION
In silico study has been successfully estimated the activity of twelve chalcone-based 1,5-benzotiazepines as potential inhibitors for breast cancer MCF7. Based on the in silico results, MA9 with binding free energy of -11.2 kcal, can interact through hydrogen bonds with amino acids Cys788 on the active site of 1T46 protein. This potentially active candidate exhibited lowest binding free energy conformation which indicated its stability during the molecular dynamic simulation. ADME profiles also proofed that its drug likeness properties. The computational results were accordance with the biological activity assay. Hence, compound MA9 was then chosen as the reference for the next stage in the drug design.
ACKNOWLEDGMENT
This research was supported by the Ministry of Research, Technology and Higher Education of the Republic of Indonesia, i.e. DRPM KEMENRISTEK for research scheme.
REFERENCES
Adel, Z., Hilwan, Y.T., Eni, N.R., Neni, F., and Ihsan, I. 2019. Synthesis and in silico studies of a benzenesulfonyl curcumin analogue as a new anti dengue virus type 2 (DEN2) NS2B/NS3. Indian Journal of Pharmaceutical Sciences. 30: 84-90.
Ameta, K.L., Nitu, S.R., and Biresh, K. 2013. Synthesis and preliminary evaluation of novel 1, 5-benzothiazepine derivates as anti-lung cancer agents. International Journal of Pharmaceutics. 3: 328-333.
Duan, L., Guo, X., Cong, Y., Feng, G., Li, John, Y., and Zhang, Z.H. 2019. Accelerated molecular dynamics simulation for helical proteins folding in explicit water. Frontiers in Chemistry. 7: 540-558.
Gaikwad, S., Venkat, S., and Kishan, L. 2013. Synthesis and antimicrobial study of novel 2, 3-dihydro-4-(naphtho [2,1-b] furan-2yl)-2-substitued [1,5] benzothiazepines. Journal of Chemical, Biological and Physical Sciences. 3: 936-940.
Garg, N., Chandra, T., Archana., Jain, A.B., and Kumar, A. 2010. Synthesis and evaluation of some new substituted benzothiazepine and benzoxazepine derivates as anticonvulsant agents. European Journal of Medicinal Chemistry. 45: 1529-1535.
Jasril, Ihsan, I., Adel, Z., Hilwan, Y.T., and Neni, F. 2017. New fluorinated chalcone and pyrazoline analogs: Synthesis, docking, and molecular dynamic studies as anticancer agents. Thai Journal of Pharmaceutical Sciences. 41: 1-6.
Kurokawa, J., Adachi-Akahane, S., and Nagao, T. 1997. 1, 5-benzothiazepine binding domain is located on the extracellular side of the cardiac l-type ca2+ channel. The American Society for Pharmacology and Experimental Therapeutics. 51: 262-268.
Lelita, R., Rahmat, G., and Winni, A. 2017. Studi Docking molekular senyawa kuersetin, kalkon, dan turunannya sebagai inhibitor sel kanker payudara MCF-7 (Michigan cancer foundation-7). Jurnal Atomik. 01: 190-196.
Lipinski, C.A., Lombardo, F., Dominy, B.W., and Feeney, P.J. 2012. Advanced drug delivery reviews, Supplement. 64: 4–17.
Mannhold, R., Kubinyi, H., and Folkers, G. 2006. Pharmacophore and pharmacophore searches. Weinhem: Wiley-VCH. 3.
Narendran, K., and Nanthini, R. 2014. A review on biological potential of chalcone hybrids. Indo American Journal of Pharmaceutical Research. 4: 3011-3022.
Neni, F., Ihsan, I., Rahma, D., Tiara, T.A., Fri, M., and Adel, Z.A. 2020 Computational approach to drug discovery: search for chalcone analogues as the potential candidates for anti colorectal cancer (HT29). Walailak Journal of Science and Technology. 12: 64-74.
Noval, H., Riska, P., Daniel, S., Neni, F., and Adel, Z. 2020. Synthesis, antiproliferative activity and molecular docking studies of 1, 3, 5-triaryl pyrazole compound as estrogen α receptor inhibitor targeting mcf-7 cells line 2020. Molekul. 15: 18 – 25.
Rebbeca, L., Siegel., and Kimberly, D.M. 2018. Cancer statistics 2017. CA: A Cancer Journal for Clinicians. 1-24.
Trott, O., Olson, A.J. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, Journal of Computational Chemistry. 31: 455-461.
Vyawahare, D., Ghodke, M., and Nikalje, A.P. 2010. Green synthesis and pharmacological screening of novel 1,5-benzothiazepines as CNS agents. International Journal of Pharmacy and Pharmaceutical Sciences. 2: 27-29.
Yamashita, F. and Hashida, M. 2004. Drug Metab. Pharmacokin. 19: 327–338.
Yaeghoobi, M.Y. 2012. Synthesis of chalcone based-six and seven membered heterocyclic compounds and their biological activities against H1N1 virus. Thesis. University of Malaya. 121-133.
OPEN access freely available online
Chiang Mai University Journal of Natural Sciences [ISSN 16851994]
Chiang Mai University, Thailand
https://cmuj.cmu.ac.th
Neni Frimayanti1,*, Marzieh Yaeghoobi2, Ihsan Ikhtiarudin1, Dhea Rizki Wannisyah Putri1, Hamid Namavar2 and Fatemeh S.Bitaraf3
1 Sekolah Tinggi Ilmu Farmasi (STIFAR) Riau, Jalan Kamboja, Pekanbaru, 28293, Indonesia
2 School of Medicine, Shahroud University of Medical Sciences, Shahroud, Iran
3 Department of Medical Biotechnology, School of Medicine, Shahroud University of Medical Sciences, Shahroud, Iran
Corresponding author: Neni Frimayanti, E-mail: nenifrimayanti@gmail.com
Total Article Views
Editor: Wasu Pathom-aree,
Chiang Mai University, Thailand
Article history:
Received: May 17, 2020;
Revised: September 30, 2020;
Accepted: October 22, 2020