 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Selection of Ankyrin Targeting HIV-1 Matrix and Identification of Its Binding Domain
Weeraya Thongkum, Kanokwan Samerjai, Somphot Saoin, Tanchanok Wisitponchai, Sudarat Hadpech, Vannajan Sanghiran Lee, Theam Soon Lim, and Chatchai TayapiwatanaPublished Date : 2018-10-01
DOI : https://doi.org/10.12982/CMUJNS.2018.0024
Journal Issues : Number 4 , October-December 2018
ABSTRACT
Ankyrin repeat protein is a novel class of non-antibody binding protein that can be applied as an alternative antiretroviral agent. Engineered ankyrin targeting the HIV-1 matrix (MA) would be a promising agent to interfere with HIV replication, since MA plays a major role in multiple processes of the viral life cycle. In this study, MA-specific ankyrin (AnkGAGG31) was isolated from an artificial ankyrin library using a semi-automated selection process with biotinylated MA-streptavidin magnetic beads. The AnkGAGG31-recognition site on MA was determined using both indirect and competitive ELISAs with overlapping MA tri-helical fragments and pentadecapeptides. The AnkGAGG31 showed the highest binding signal to the MA-fragments covering helices 2-3-4 and peptides corresponding to helix 2 (residues 25-43), which were found as the target epitope. This finding was further analyzed by molecular modeling and docking. The rational models of AnkGAGG31-MA complex indicated that the strong binding interaction was shown on helix 2 at key residues K27MA, K30M, and K32MA. Taken together, the identification of the binding domain on the MA target improves our understanding of the AnkGAGG31-MA interaction and provides the information necessary to design innovative protein targeting of the MA protein.
Keywords: Ankyrin, Binding domain, HIV-1 matrix, MA protein, Phage-displayed panning
INTRODUCTION
Human immunodeficiency virus type-1 matrix (HIV-1 MA) is one of the major domains of the Gag polyprotein precursor that is involved in multiple steps of the viral life cycle, including viral assembly and maturation (Scarlata and Carter, 2003; Bukrinskaya, 2007; Sundquist and Kräusslic, 2012; Bell and Lever, 2013). The MA and nucleocapsid simultaneously interact with viral genomic RNA, which leads to Gag-Gag polymerization; this drives the assembly and targeting of Gag multimerization to the plasma membrane via the N-terminal myristoylated MA domain. In addition, the released immature virions permit infectious maturation through sequential proteolytic processing of Gag polyprotein, such as cleavage at the matrix-capsid (CA) junction (Scarlata and Carter, 2003; Sundquist and Kräusslic, 2012; Bell and Lever, 2013). Therefore, HIV-1 MA is an interesting target to try to inhibit viral replication, since inhibiting MA-function might disrupt the production of infectious viral progeny at the early stages of assembly (Muriaux et al., 2004; Sticht et al., 2005). Although the current treatment of HIV infection (known as highly active antiretroviral therapy, or HAART) can suppress the viral levels in patients, it is not effective in all cases (Sticht et al., 2005; Paredes and Clotet, 2010; Arts and Hazuda, 2012). In addition, HAART therapy can lead to the emergence of multidrug-resistant HIV-1 strains, a long-lived pool of latently infected cells, and adverse effects of long-term treatment (Luque et al., 2005; Paredes and Clotet, 2010; Chen et al., 2013). As a result, new treatment options continue to be pursued.
Recently, anti-AT20 antibody, or intracellular single chain Fv fragments (scFv), derived from MH-SVM33 has been proposed as a neutralizing antibody that binds to the conserved domain of MA (Sticht et al., 2005; Fiorentini et al., 2008; Tue-ngeun et al., 2013). Although it was a promising candidate as a new antiretroviral agent, it still lacks bioavailability data because of its limited solubility and binding affinity in an intracellular expression system. Consequently, numerous alternative protein scaffold-based agents have been proposed and developed to overcome the limitations of therapeutic antibodies (Tamaskovic et al., 2012; Sawyer et al., 2013). Since alternative non-antibody frameworks are highly soluble and stable, they offer an attractive substitute for antibodies or their derivatives (Binz et al., 2005; Gilbreth and Koide, 2012).
Ankyrin repeat protein is one important protein-protein interaction motif that can be modified for target protein binding with high specificity and high affinity (Tamaskovic et al., 2012; Plückthun, 2015). Ankyrin is highly soluble and stable, even in the intracellular reducing environment; thus allowing it to be expressed with high yields in bacteria for cost-efficient production (Binz et al., 2003; Tamaskovic et al., 2012; Plückthun, 2015). Artificial ankyrin binders have been successfully developed as a blocking agent in the binding of viral gp120 to its receptor CD4 (Schweizer et al., 2008), or for inhibiting HIV-1 CA (referred to as AnkGAG1D4); they could serve as a novel intracellular inhibitor of viral assembly (Nangola et al., 2012; Praditwongwan et al., 2014; Khamaikawin et al., 2015). No research to date has reported on ankyrin that specifically binds to the MA protein of HIV.
The aim of this study was to select ankyrin binders targeting HIV-1 MA and to identify the binding site of the selected ankyrin on the MA protein. The MA-specific ankyrin was isolated using a semi-automated panning process with a phage-display library against a biotinylated MA coupled to streptavidin magnetic beads. The in vivo biotinylation preparation allows for rapid and specific conjugation of MA protein with biotin carboxyl carrier protein (BCCP) tag (Tayapiwatana et al., 2006; Orrapin and Intorasoot, 2014). The semi-automated panning process allows for a more efficient and less time-consuming process compared to conventional microtiter plate panning (Konthur et al., 2010; Chin et al., 2016). The determinations of binding site on MA were analyzed using MA tri-helical fragments and pentadecapeptides scanning by indirect and competitive ELISAs, respectively. In addition, a computational approach was used to propose a plausible model of ankyrin-MA interaction. The docking complexes were generated by ZDOCK and RDOCK protocols. Subsequently, 10 docking complexes relying on ZRANK that showed the highest efficiency among ZDOCK parameters were chosen for characterization. The characterization of the selected ankyrin using experimental and computational analysis could provide insights to the molecular mechanisms of ankyrin targeting HIV-1 MA. The information can potentially be used to design new antivirals as MA-inhibitors or to interfere in HIV assembly.
MATERIALS AND METHODS
Bacterial strains and vectors
The pNL4-3 plasmid obtained from the NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH, MD) was used as a template for amplification of HIV-1 MA fragment genes. The recombinant plasmids were transformed into a cloning host, Escherichia coli strain XL1-blue (Stratagene, La Jolla, CA), for propagation and screening of hosts containing the plasmid of interest. E.coli strain Origami B (Novagen, Madison, WI) was used as the host for expression of biotinylated protein-BCCP tag from the pAK400cB vector, which was a generous gift from Ville Santala (University of Turku, Finland) (Tayapiwatana et al., 2006). The expression vector pQE30 (Qiagen, Hilden, Germany) was used for production of 6xHis-tagged recombinant ankyrin in E.coli strain M15 [pREP4] cells (Qiagen, Hilden, Germany).
Production of non-chemical conjugation of biotinylated H6-MA-BCCP
The DNA fragments encoding the overlapping α-helix MA region were amplified from the pNL4-3 plasmid using HotStar Hifidelity DNA polymerase (Qiagen, Hilden, Germany) and a following pair of primers, as shown in Table 1. PCR fragments were treated with NdeI and EcoRI (Thermo Scientific, Waltham, MA) and then ligated to the pAK400cb-BCCP. The recombinant plasmids were transformed into XL1-Blue and selected on LB agar containing tetracycline, chloramphenicol, and kanamycin. The correctness of clones was identified using a standard DNA sequencing method. The purified pAK400cb carrying α-helix MA fragment gene was transformed into E. coli strain Origami B for protein expression.
E. coli strain Origami B harboring the pAK400cb-MA fragments were cultured in terrific broth containing glucose (0.05% w/v), kanamycin (15 μg/mL), tetracycline (12.5 μg/mL), chloramphenicol (25 μg/mL), and 4 μM D-biotin (Sigma-Aldrich, St. Louis, MO). The culture was incubated at 37°C with shaking at 200 rpm, until OD600 nm reached 0.8. Induction of protein expression was performed by addition of 0.1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) and incubated at 25°C for 18 hours with shaking. After incubation, bacterial cells were collected by centrifugation at 5,000×g, 4ºC for 30 minutes. The cell pellets were resuspended in PBS, pH 7.4 including protease inhibitor (Millipore, Billerica, MA). The pellet was then lysed by three cycles of freezing-thawing method and sonication. After centrifugation, the supernatant was collected.
Table 1. The primers used to generate the H6-MA fragments
Preparation of MA-magnetic beads
Biotinylated MA full length protein (100–200 μg) was dissolved in 1 mL PBS. One milligram of Dynabeads M-280 Streptavidin magnetic nanoparticles (Invitrogen, Waltham, MA) were resuspended gently in solution of biotinylated MA and incubated for one hour on a rotator at room temperature. The beads were harvested by a magnet and the supernatant removed. Then, MA-magnetic beads were washed three times with 1.5 mL of PBS with 0.1% Tween-20. Finally, the MA-magnetic beads were removed from the magnet and resuspended in 200 μl of PBS.
Preparation of phage displayed artificial ankyrin library
The ankyrin library was as previously described (Nangola et al., 2012). The ankyrin library stock was inoculated in a 1:100 ratio with 2YT broth containing ampicillin and 2% glucose at 37°C with shaking until OD600 nm reached 0.4-0.5. The culture was infected with helper phage M13K07 (New England Biolabs, Ipswich, MA) and incubated statically for 30 minutes at 37°C. The cell pellet was collected by centrifugation at 1,200×g for 10 minutes. New broth containing ampicillin, kanamycin, and IPTG was used to resuspend the pellets and incubated overnight at 30°C with shaking. Then, the supernatant was collected and precipitated with PEG/NaCl (20% polyethylene glycol 8,000, 2.5 M NaCl) on ice for one hour and centrifuged to collect the pellet. Lastly, the pellet was resuspended in 1 mL of PBS.
Phage-displayed library panning with magnetic bead
The panning selection was carried out with an automated magnetic-particle processor, KingFisher (Thermo Scientific, Waltham, MA) (Konthur et al., 2010; Chin et al., 2016). Briefly, phage-displayed ankyrin library was preincubated with PTM (PBS + 0.1% Tween 20 +2% skimmed milk) for one hour. The pre-incubated phage-displayed library was mixed with the MA-magnetic beads for one hour with shaking. For the first panning round, the beads were washed twice with PBS containing 0.1% Tween 20 and once with rod-shaped magnets at medium speed. The washing frequency was increased with subsequent rounds of panning. The beads were then transferred to a new plate by the magnets and released by resuspension in 300 μL of PBS. To screen and select the clones, the MA-specific bound phages were detected with horseradish peroxidase (HRP)-conjugated anti-M13 (dilution 1:5,000) and ABTS solution (GE Healthcare Life Sciences, Piscataway, NJ). Absorbance of each well was measured at 405 nm. For phage rescue, E. coli TG1 cells with OD600 nm 0.4-0.5 were added and incubated for 30 minutes at 37°C. Subsequently, 2YT broth containing ampicillin and 2% glucose was added and then incubated at 37°C for two hours with shaking. Helper phage M13K07 was added and incubated for another 30 minutes at 37°C. After incubation, the pellets were collected by centrifugation, resuspended in a new 2YT broth, and incubated overnight at 30°C. The supernatant was collected and used for a consecutive round of panning or selection of the monoclonal anykyrin that react to HIV-1 MA protein.
Expression and purification of soluble H6-ankyrin
The phagemid pHDiExDsbA-ankyrin from panning selection was treated with NotI and HindIII (Thermo Scientific, Waltham, MA), and the excised ankyrin module subsequently cloned into the expression vector pQE30. The correctness of clones was verified by a standard DNA sequencing method (1st BASE Pte Ltd, Singapore). The constructed plasmid was transformed into E. coli strain M15[pREP4], which carries the deletion in the lacZ gene to disrupt the formation of functional β-galactosidase gene. This strain additionally contains lac repressor (lacIQ gene) on the pREP4 plasmid to operate directly on the expression plasmid to repress transcription from a T5 promoter. The bacteria harboring the pQE30-ankyrin plasmid was grown in terrific broth containing kanamycin (25 μg/mL) and ampicillin (100 μg/mL). Induction of ankyrin expression was performed by addition of 0.1 mM IPTG and incubated at 30°C for 18 hours with shaking. After incubation, bacterial cells were collected following the protocol as detailed above. The soluble H6-ankyrin was purified by affinity chromatography on a HisTrap affinity column and ÄKTA primeTM plus system (GE Healthcare Life Sciences, Piscataway, NJ).
Screening of ankyrin reactivity towards HIV-1 MA protein
To assay whether purified ankyrin specifically bound to MA protein, an ELISA was performed. A microtiter plate was coated with biotinylated MA-BCCP, H6-MACA, or H6-CA. The purified H6-MACA and H6-CA was produced in baculovirus-infected cells, as described in a previous study (Nangola et al., 2012). The coated plate was blocked with 2% skimmed milk in PBS and washed with 0.05% Tween-20 in PBS. The purified ankyrin (10 μg/mL) was added to each well and incubated for one hour. Then, a binding reaction was detected with rabbit anti-ankyrin polyclonal antibody (pAb) (dilution 1:30,000) followed by goat anti-rabbit IgG-HRP at a dilution of 1:5,000 (KPL, Gaithersburg, MD). Then, TMB substrate was added into each well of the plate followed by incubation for 15 minutes at 25°C. Finally, the reaction was stopped by adding 1 N HCl and the absorbance was measured at OD450 nm.
Western immunoblotting
The H6-ankyrin was separated by SDS-PAGE under reducing condition and blotted on a polyvinylidene fluoride (PVDF) membrane. The PVDF membranes were incubated in blocking buffer (2% skimmed milk in PBS) and washed with PBS containing 0.05% tween 20. The purified ankyrin was detected with rabbit anti-ankyrin pAb (dilution 1:30,000) and anti-His tag monoclonal antibody (mAb). The primary mouse antibody was followed by goat antimouse immunoglobulins-HRP (dilution 1:3,000) (KPL, Gaithersburg, MD). Goat anti-rabbit IgG-HRP (KPL,Gaithersburg, MD) at a dilution of 1:5,000 was added to detect the rabbit anti-ankyrin pAb. After the last wash step, the reaction was visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA) and exposed on X-ray film (Agfa HealthCare, Belgium).
Determining the binding domain on MA recognized by AnkGAGG31
Indirect ELISA. Microtiter plate was coated with 10 μg/mL of avidin, H6-MACA, or H6-CA, and left overnight at 4°C. The coated plate was blocked with 2% skimmed milk in PBS, and then washed five times with high stringency washing buffer (1% Triton X100 (v/v) and 550 mM NaCl prepared in PBS, pH7.4) (Saoin et al., 2017). Fifty microliters each of bacterial extract (1 mg/mL) comprised of biotinylated H6-MA-fragments, survivin-BCCP, or Origami B lysate were individually added into each avidin-coated well and incubated for one hour at 25°C. After washing, 50 μL of AnkGAGG31 (10 μg/mL) was added into each well and incubated for one hour. The plate was washed five times before adding 50 μL of rabbit antiankyrin pAb (dilution 1:30,000). The reaction was detected by goat anti-rabbit IgG-HRP at a dilution of 1:5,000 and TMB substrate (KPL, Gaithersburg, MD). Finally, the absorbance of each well was measured at OD450 nm.
Competitive ELISA. The overlapping peptides covering the entire MA plus the N-terminal 13 residue of CA were used as competitors for binding of H6-MACA to the immobilized AnkGAGG31. The MA peptides consisted of a bacterial extract protein and a panel of synthetic pentadecapeptide (obtained through the NIH AIDS Reagent Program: HIV-1 Consensus A Gag Peptides - Complete Set, Cat# 8116, Lot# 1). The synthetic consensus peptides are 15-mers with 11 amino-acid overlapping sequences, which referred to F1 to F34. The plate was coated with 50 μL of purified AnkGAGG31 (final concentration of 10 μg/mL) and incubated overnight at 4°C. Next, ELISA plate was washed with the high stringency washing buffer and then blocked with PBS plus 1% skimmed milk. H6-MACA (concentration 20 μg/mL) solution was mixed in 1:1 ratio with the solution of each competitor: the biotinylated proteins (500 μg/mL), the synthetic pentadecapeptides (60 μM), AnkGAGG31 (20 μg/mL) used as the positive control, H6-CA (20 μg/mL), or origami B lysate (500 μg/mL) used as the negative control. Subsequently, 50 μL of the mixed solutions were individually added into each well and incubated for one hour at 25°C. Next, unbound proteins were removed by washing five times prior to the addition of mouse anti-CA mAb (5 μg/mL) (a kind gift from Watchara Kasinrerk, Chiang Mai University, Thailand). After incubating for one hour and then washing, 50 μL of HRP-labeled goat anti-mouse immunoglobulins (dilution of 1:3,000) (KPL, Gaithersburg, MD) were added to the wells and incubated for one hour. The plate was washed again and TMB substrate was added (KPL, Gaithersburg, MD). The colorimetric signal was measured at OD450 nm.
Homology modeling
Homology modeling was used to generate a three-dimensional structural model of the ankyrin targeting HIV-1 MA, following the homology protocol in Discovery Studio (DS) 2.5 software described elsewhere (Reddy et al., 2012; Vyas et al., 2012). The model was built based on three dimensional (3D) structures of AnkGAG1D4 (PDB ID: 4HLL) and DARPin-8.4 (PDB ID: 2Y1L) from RCSB PDB (http://www.rcsb.org/pdb/home/home.do). The AnkGAG1D4 sequence showed 84.42% sequence identity to N-cap, 2nd-4th internal repeats, and C-cap of AnkGAGG31; meanwhile, DARPin-8.4 had 79.87% sequence identity to N-cap, 1st-3rd internal repeats, and C-cap. Five homology structures were generated by the simulated annealing. The structure with the highest score of probability density function (PDF) was selected. The stereochemical quality of homology models was evaluated using Ramachandran plot and a potential energy of minimized model.
Molecular docking of AnkGAGG31-MA complex
The AnkGAGG31 homology structure and the crystal structure of HIV-1 MA from RCSB PDB (PDB ID: 1L6N, residues 10-121) were used for generating the docking complexes in DS 2.5 software. The 3D structure of AnkGAGG31 and MA were prepared by energy minimization with CHARMm force field and without incorporating solvent and salt concentrations. The algorithm for minimization began using 5,000-step energy minimization with 0.1 Å of rootmean-square deviation (RMSD) and Conjugate Gradient (CONJ) (Chen et al., 2003; Wiehe et al., 2008; Wisitponchai et al., 2017). Following ZDOCK protocol, the variable residues on each repeat of AnkGAGG31 were specified as binding residues, and atoms of MA within the region of binding interface at a distance cutoff of 10 Å were selected (Halperin et al., 2002; Wiehe et al., 2008; Wisitponchai et al., 2013). From 54,000 docking complexes, 2,000 complexes with the highest ZDock score and binding interface filter were selected. The ZRank score and cluster were calculated on 2,000 docking poses using a RMSD cut-off and interface cut-off of 6.0 and 9.0, respectively. Any poses having RMSD or interface region less than the cut-off values were grouped in the same cluster. The top-10 lowest value ZRank score poses were identified as candidates. The candidates were typed with CHARMm polar H force field, followed by refining in the refinement docked proteins (RDOCK) protocol with dielectric constant at 4.0 (Li et al., 2003; Pierce and Weng, 2008). Analyzing the complexes of AnkGAGG31-MA, the binding interface and interaction bond was calculated. The intermolecular neighbor within 5 Å was identified as the binding interface. The hydrogen was determined by the distance threshold of 2.5 Å and the donor proton-acceptor angles. The maximum criteria of the D-H-A angle, X-D-A angle, H-A-Y angle, and D-A-Y angle were defined at 180 degrees. The van der Waals (vdW) and electrostatic interaction energy were typed with CHARMm force field, and the energy value was calculated.
RESULTS
Selection of MA-ankyrin binders using MA-magnetic beads
The biotinylated H6-MA-BCCP was produced from E. coli Origami B stain harboring pAK400cb-MA supplemented with D-biotin. The in vivo expression of non-chemical conjugation of biotinylated MA was coated with streptavidin magnetic beads. This protein was assayed by Western immunoblotting. The results demonstrated that the specific band of biotinylated MA-BCCP approximately at 35 kDa was observed using anti-MA mAb and HRP-conjugated anti-biotin mAb (data not shown). As a consequence, the MA-coated streptavidin beads were utilized for phage-displayed library panning, which was performed and analyzed by phage ELISA. After five rounds of panning, the enrichment of pooled phagedisplayed ankyrin targeting MA was detected (Figure 1A). The individual eluted phages from the fifth round of panning were screened using phage ELISA. Interestingly, one reactive ankyrin binder showed high positive signal with MA antigen, as shown in Figure 1B. This new ankyrin recognizing HIV-1 MA, named AnkGAGG31, was successfully isolated from the phage-displayed library. The specificity of AnkGAGG31 with MA was subsequently evaluated by indirect ELISA. As shown in Figure 1C, the AnkGAGG31 was able to bind to H6-MA-BCCP and H6-MACA, whereas no signal could be observed in H6-CA. This result indicated that the AnkGAGG31 could specifically bind to MA protein in the conformational Gag polyprotein (H6-MACA) and the truncated Gag polyprotein (H6-MA-BCCP).
Characterization of AnkGAGG31
DNA sequencing revealed that the H6-AnkGAGG31 protein contained 202 amino acids comprised of four internal repeats flanked by N-terminal and C-terminal caps, each containing different types of amino acids at the six assigned positions for variable residues (Figure 2A). The rigid framework of individual repeat was conserved, which is similar to the consensus sequence of the ankyrin library. Meanwhile, most variable residues of each repeat consisted of polar amino acids. The purified tetramodular AnkGAGG31 was examined by Western immunoblotting using anti-His-tag mAb and rabbit anti-ankyrin pAb. The result showed that AnkGAGG31 migrated, with an apparent molecular weight of 22 kDa (Figure 2B), consistent with the theoretical mass for a protein of 202 amino acid residues. Furthermore, the 3D structure of AnkGAGG31 (Figure 2C) was constructed by homology modeling, using AnkGAG1D4 (PDB ID: 4HLL, 84.42% sequence identity) and DARPin-8.4 (PDB ID: 2Y1L, 79.87% sequence identity) as the templates. The quality of AnkGAGG31 was assessed by 92.10% of residues that were found in the core region of the Ramachandran plot (Figure 2D) and showed -11,717.02 kcal/mol of potential energy. This homologous structure of AnkGAGG31 in its lowest energy conformation was selected to generate protein-protein docking simulation with MA protein.
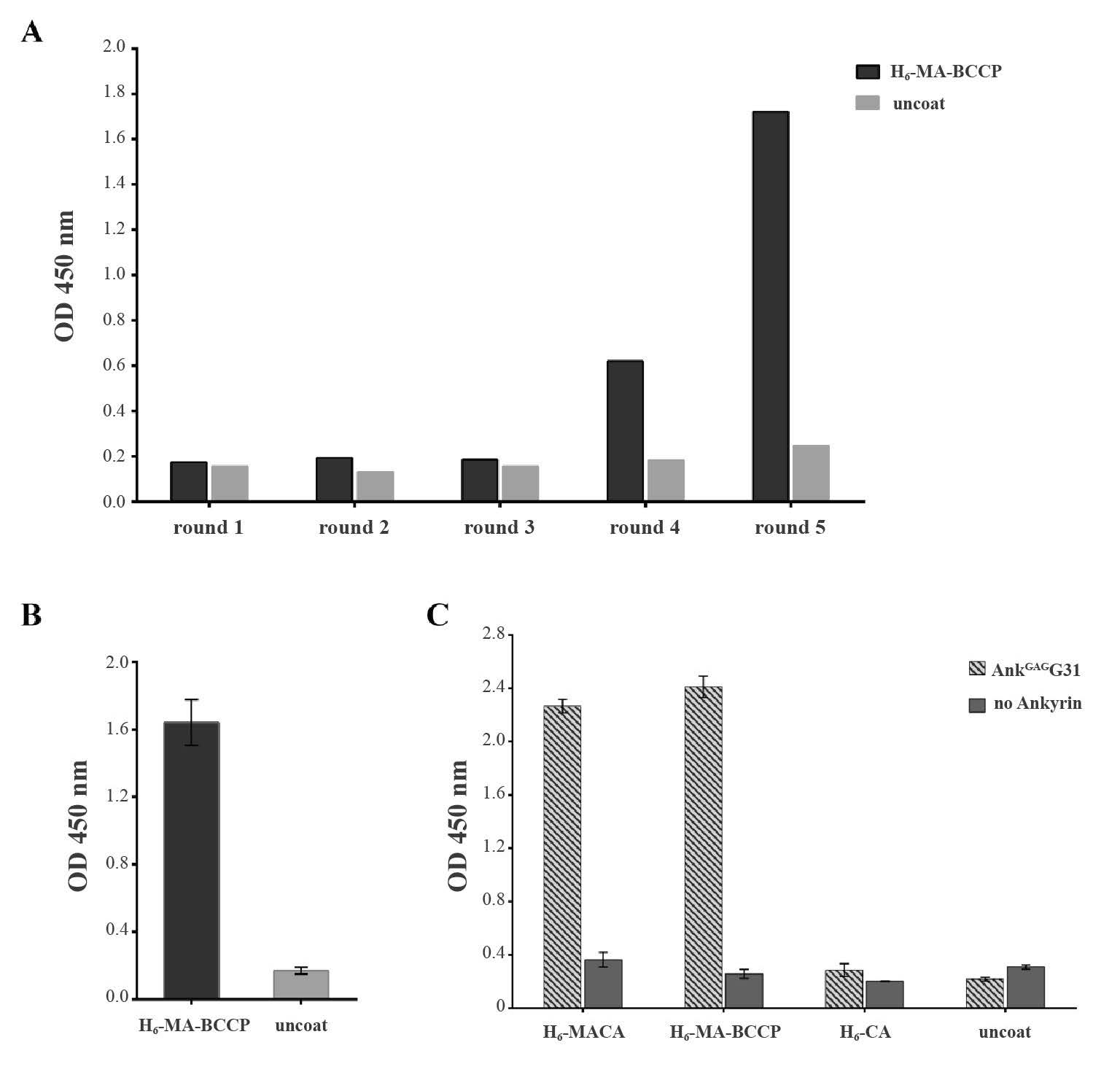
Figure 1. Selection of ankyrin targeting HIV-1 MA. (A) Binding activity of the pooled phage-displayed ankyrin from five rounds of panning with MA-magnetic bead. (B) Binding activity of the phage clone, named AnkGAGG31, isolated from the fifth round phage-displayed library panning. (C) The reactivity of AnkGAGG31 with H6-MACA, H6-MA-BCCP, and H6-CA analyzed by indirect ELISA and using anti-ankyrin pAb for detection.
Note: Error bars represent the mean ± SD of three independent experiments performed in triplicate.
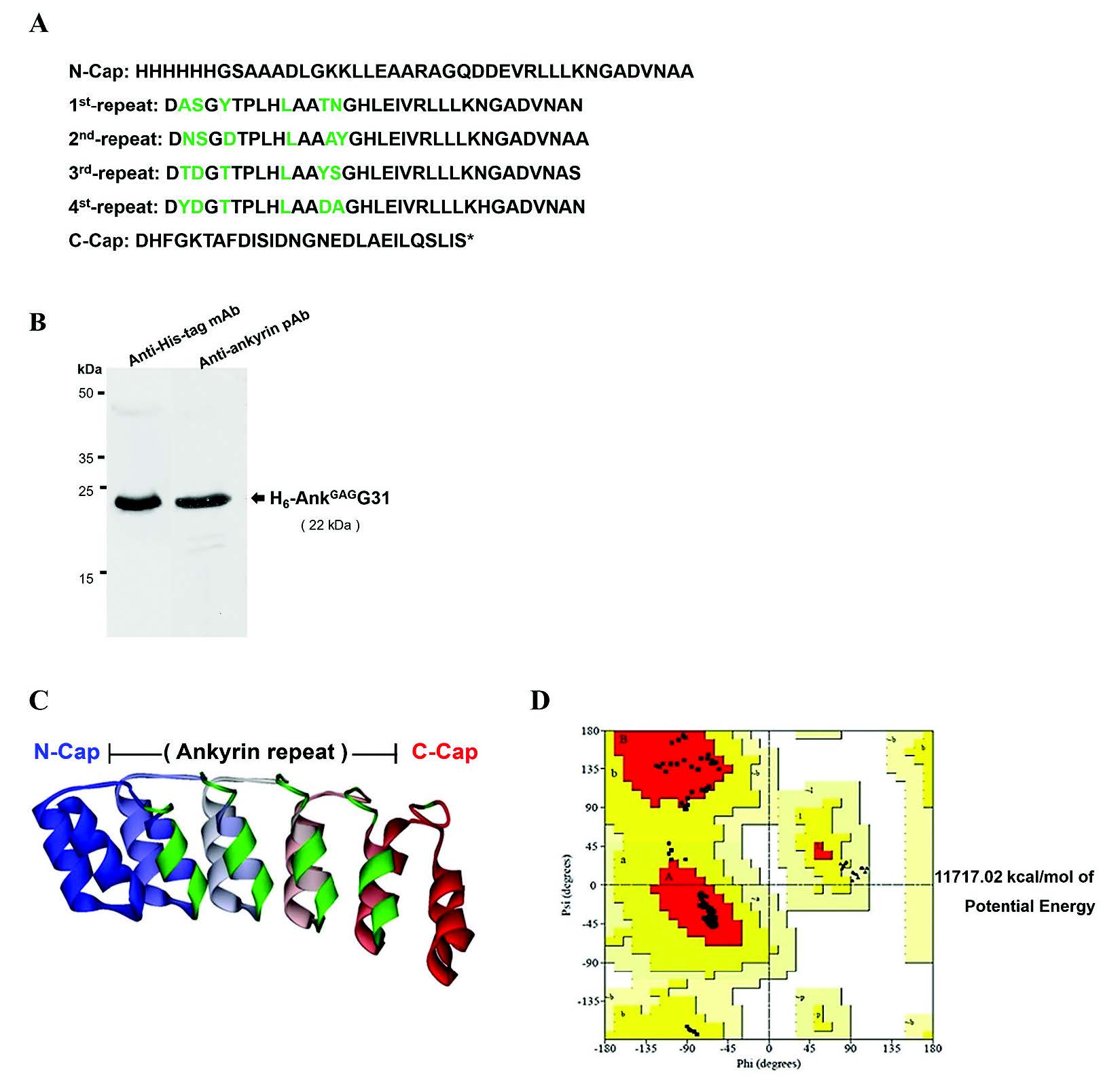
Figure 2. Characterization of AnkGAGG31. (A) Amino acids sequence of AnkGAGG31. The variable residues on each ankyrin repeat are highlighted in light gray. (B) Western immunoblotting analysis of purified H6-AnkGAGG31 protein (10 μg/lane) using anti-His-tag mAb and anti-ankyrin pAb. (C) Three-dimensional structural model of AnkGAGG31 showing four internal repeats, which is presented using a gray scale of gray to white in ribbon style. The black color represents variable residues. (D) The Ramachandran plot of minimizing structures of the AnkGAGG31 homology model shows the amino acids in core region, allowed region, generously allowed region, and disallowed region as dark gray (d), gray (g), light gray (l), and white, respectively.
Determination of AnkGAGG31–recognition site on HIV-1 MA protein
To identify the binding domain of AnkGAGG31 on MA, both indirect ELISA and competitive ELISAs were performed using the overlapping tri-helical MA fragments and a panel of synthetic pentadecapeptide covering the HIV-1 MA domain. Since the three-dimensional structure of MA consists of five α-helical regions (Fiorentini et al., 2006; Bukrinskaya, 2007), design of the MA fragments was associated with overlapping proportions of α-helix, including: MA-F123 (α-helices 1-2-3 or 1M-Q69), MA-F234 (α-helices 2-3-4 or 19I- R91), MA-F345 (α-helices 3-4-5 or 46V-Q145), and H6-MA (the whole MA domainconnecting 13 residues of capsid or 1M-Q145).
First, the binding activity of MA-specific ankyrin was examined by indirect ELISA. The microtiter plate was pre-coated with the tri-helical MA-fragment; MA-F123-BCCP, MA-F234-BCCP, and MA-F345-BCCP, respectively. Interaction of AnkGAGG31 to the immobilized overlapping tri-helical MA-fragments was monitored. As shown in Figure 3A, AnkGAGG31 produced high binding signals to the MA-F234-BCCP, and MA-full length-BCCP and H6-MACA fragments (the whole MA domain served as the positive control). In contrast, the fragments of MA-F123-BCCP and MA-F345-BCCP presented low binding signals, similar to the signal-binding of the negative control. This result implied that AnkGAGG31 specifically bound to MA-F234-BCCH, which referred to helices-2-3-4 of the MA domain.
Second, competitive ELISA was used to ensure the binding activity of the AnkGAGG31 to its target. AnkGAGG31 was used as the immobilized ligand and H6-MACA was used as the soluble ligand. The competitors consisted of the overlapping biotinylated tri-helical MAfragments and the synthetic pentadecapeptides. The results indicated that the three competitors –MA-BCCP, F234-BCCP, and F123-BCCP – were able to compete with the H6-MACA to bind to immobilized AnkGAGG31. The reduction of the binding signals were calculated to percentage of inhibition, as shown in Figure 3B (MA-BCCP = 88.7%, F234-BCCP = 81.0%, and F123-BCCP = 69.5% inhibition). AnkGAGG31, used as the self-competitor, showed 82.6% inhibition. The average percentage of inhibition using the negative reference was 47.3%. The negative control in this study consisted of Origami B lysate and biotinylated survivin. Mean percent inhibition of F123-BCCP, F234-BCCP, and MA-BCCP revealed a significant difference from the average percent inhibition of the negative reference. This result highlighted the possibility that the AnkGAGG31 was targeting the α-helix 2 or 3 of the MA domain.
Additionally, the competitors were changed from the overlapping biotinylated tri-helical MA-fragments to the overlapping pentadecapeptides, as shown in Figure 3C. Two overlapping pentadecapeptides, F7 and F8 (amino acid residues 25-43) corresponding to MAhelix 2, competed with the binding of AnkGAGG31 to H6-MACA. These two competitors had percentages of inhibition of 53.2% and 43.7%, respectively; these were significantly over the cut-off value (20.5%). Note that the cut-off value was derived from the average percentage of inhibition by the negative control, using the negative references of H6-CA (23.3%) and CA-peptide (18.5%). The overlapping pentadecapeptides that were marked as significant competitors were chosen due to their competitive effect that was greater than two standard deviations of the average percentage of inhibition by the negative reference. Taken together, the result of the indirect and competitive ELISAs suggested that the AnkGAGG31 specifically bound to the MA domain on residues of α-helix 2. Nevertheless, to ensure which helix is the AnkGAGG31-recognition site, we conducted a computational study using the homology modeling of AnkGAGG31 and molecular docking.
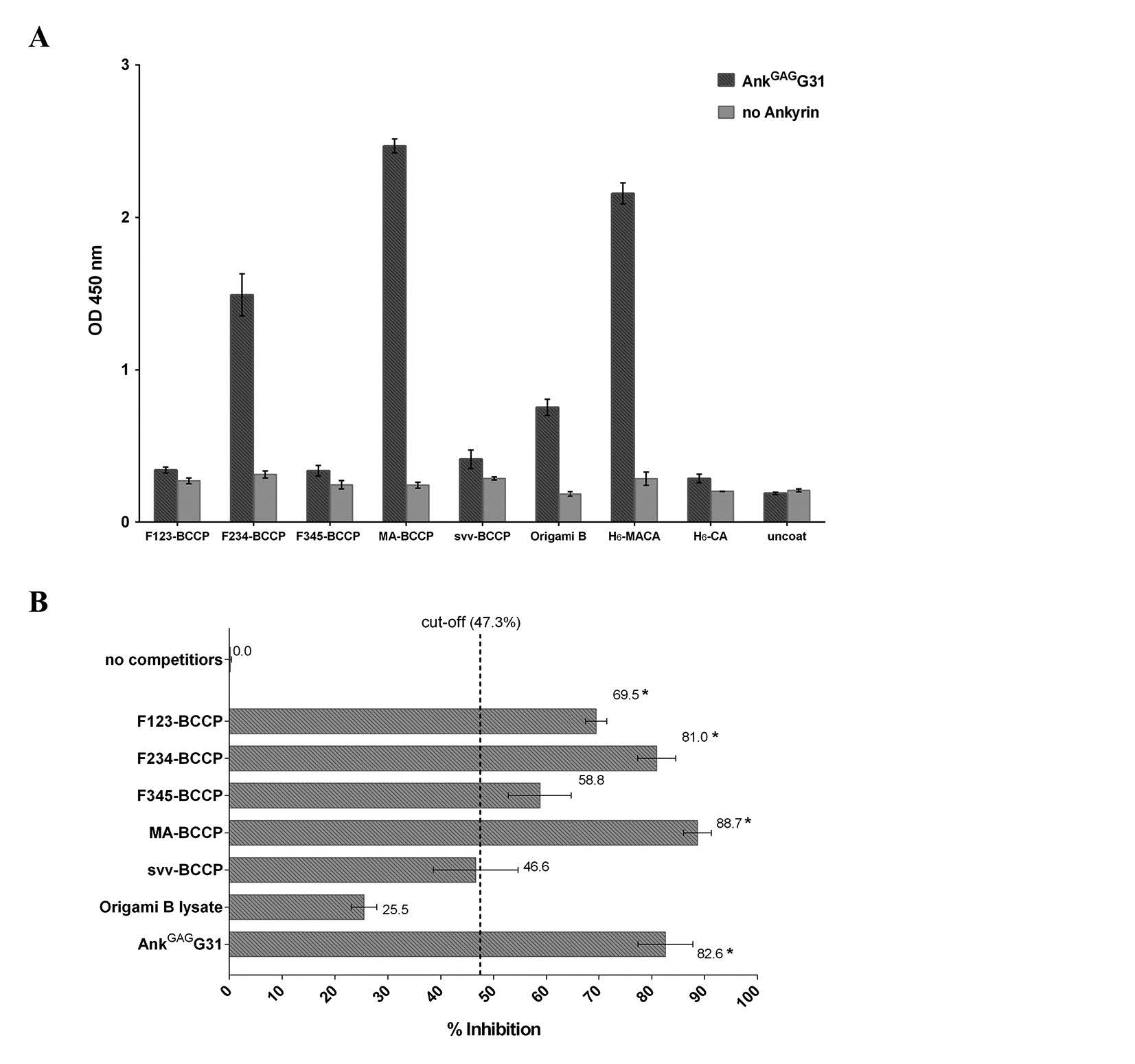
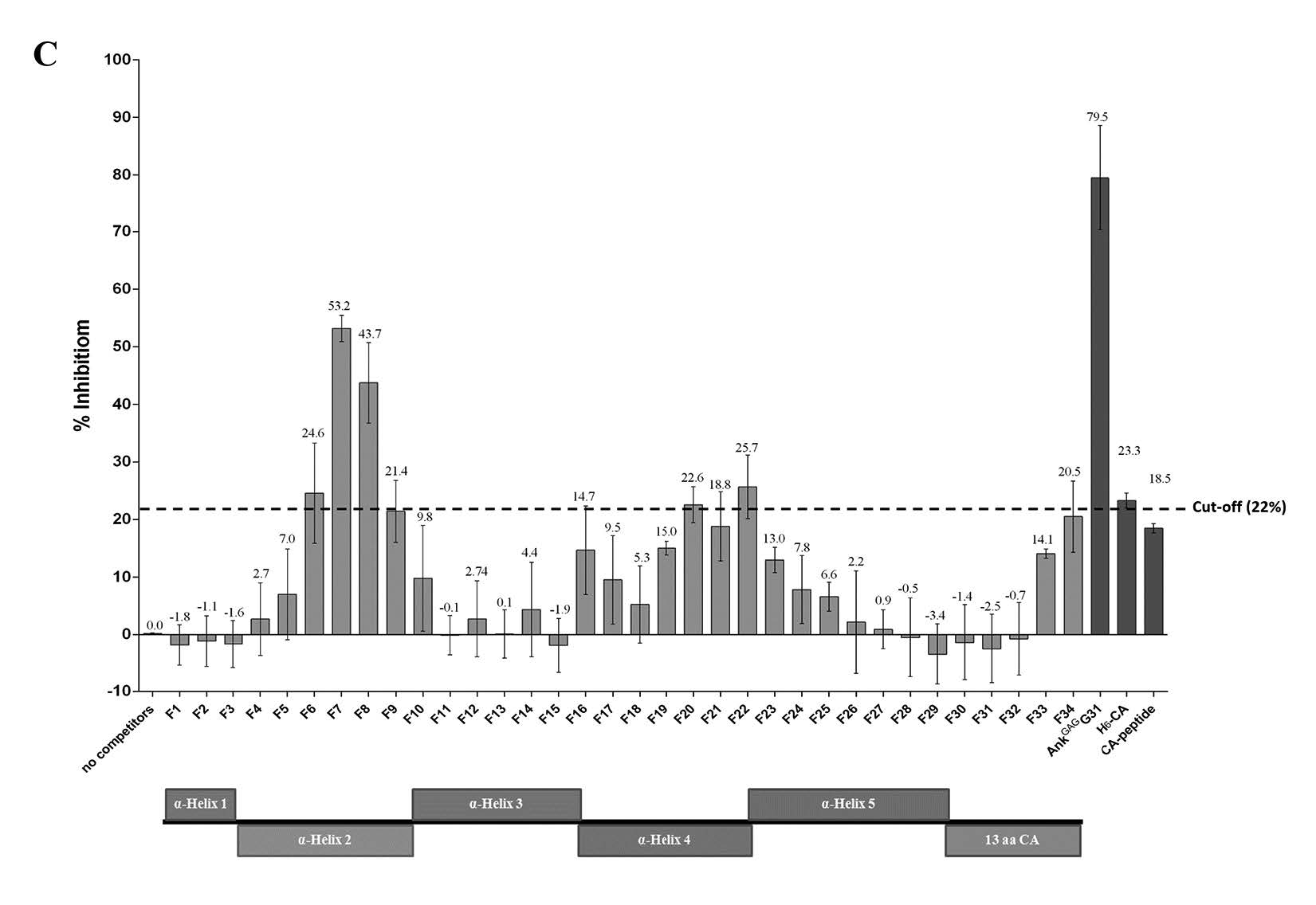
Figure 3. Determining the binding domain on MA recognized by AnkGAGG31. (A) The direct reactivity analysis of AnkGAGG31 with MA-fragments using indirect ELISA. The sample proteins-coated plate are detected with AnkGAGG31, and followed by anti-ankyrin polyclonal antibody. Data shown represent mean ± SD of results in triplicate. Analysis of AnkGAGG31 binding to H6-MACA in the presence of competing biotinylated MA-fragments (B) and pentadecapeptides (C). Overlapping pentadecapeptides are referred to as F1 to F34, relative to the regions of α-helical MA (represented as boxes). The dashed lines represent the average percent inhibition of negative control define at 47.3% and 22%, respectively.
Note: Data shown represent the mean ± S.D. of percent inhibition from the results in triplicate. * Denotes values that differ significantly from negative reference control, P < 0.05.
Docking simulation of AnkGAGG31 binding to HIV-MA
To consider the possible structures of the hypothetical complexes between AnkGAGG31 and HIV-1 MA, the reliable structures could define a hypothetical result from its low-energy conformations (Pierce and Weng, 2008; Wiehe et al., 2008; Wisitponchai et al., 2013). The homology model of AnkGAGG31 and the 3D structures of MA (PDB ID: 1L6N) were built using a docking simulation method (ZDOCK and RDOCK protocols) in DS 2.5 software. The 1,791 docking complexes (poses) of AnkGAGG31-MA were generated and the 10 lowestenergy docking poses (namely, P1-P10) ranked by ZRank score were chosen to analyze the binding interface and interaction energy. The result showed that the 10 docking complexes were represented in four conformations, named cluster 3 (C3, n=1), cluster 13 (C13, n=2), cluster 14 (C14, n=3), and cluster 24 (C24, n=4), as shown in Figure 4. Interestingly, in all four clusters, AnkGAGG31 bound to MA-helix 2 (and/or helices 1 and 5) that corresponded with the sequences of F7 and F8 from the analysis of competitive ELISA. To analyze the interface MA-residues 25-43 (belonging to pentadecapeptide F7 and F8) that specifically interacted to AnkGAGG31, the highest surface area appeared in the docking pose named P6 (1382.1 Å2), which presented in C24 (Figure 4). Similarly, almost all of the maximum values of each binding energy were shown in C24, including the highest ZRank (-130.7 kcal/mol) which presented in P1, the highest interaction energy of complex from electrostatic bond (-1,254.2 kcal/mol) which showed in P4, and the highest total interaction energy between MA residues 25-43 and AnkGAGG31 (-1,136.4 kcal/mol) which belonged to P3, as shown in Table 2. Therefore, C24 was likely the conformation that presented for the AnkGAGG31-MA interaction. Since P3 was located in the center of C24 and showed the highest total interaction energy between MA residues 25GKKKYRLKHLVWASRELER43 and AnkGAGG31, it was used as a candidate for the 10 docking poses.
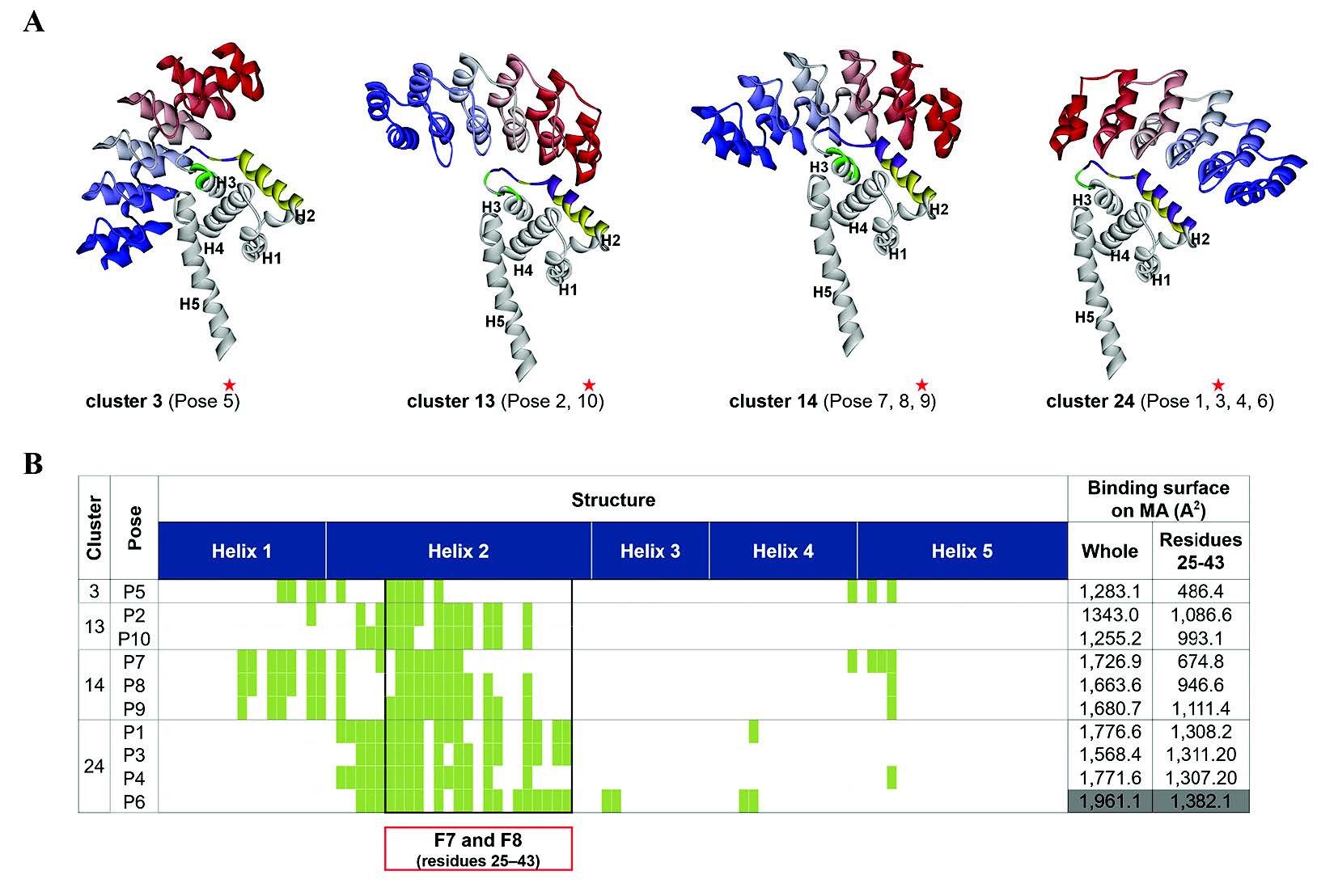
Figure 4. Characterization of the 10 lowest energy docking poses of AnkGAGG31-MA. (A) Four conformations of 10 docking complexes in each cluster (C), named C3, C13, C14, and C24. The representative model of each cluster (marked by black stars) is a center of cluster. The ribbon style in a gray scale of gray to white defines the N- to C-terminus direction of AnkGAGG31. Meanwhile, the considered residues 25-43 on a white ribbon of MA are shown in gray. The AnkGAGG31 binding interface with a 5 Å cut-off is shown in the white ribbon of MA. (B) The interface MA residues are relative to Fig. 5A and shown in linear sequence. The residues 25-43, corresponding to F7 and F8 pentadecapeptides, are presented in an area of a black square table. The binding surface on MA that interacted with AnkGAGG31 is calculated from 10 docking structures shown in the last two columns. The maximum value of binding interface is highlighted in gray.
Table 2. Interaction energy of the 10 lowest energy docking poses of AnkGAGG31-MA.
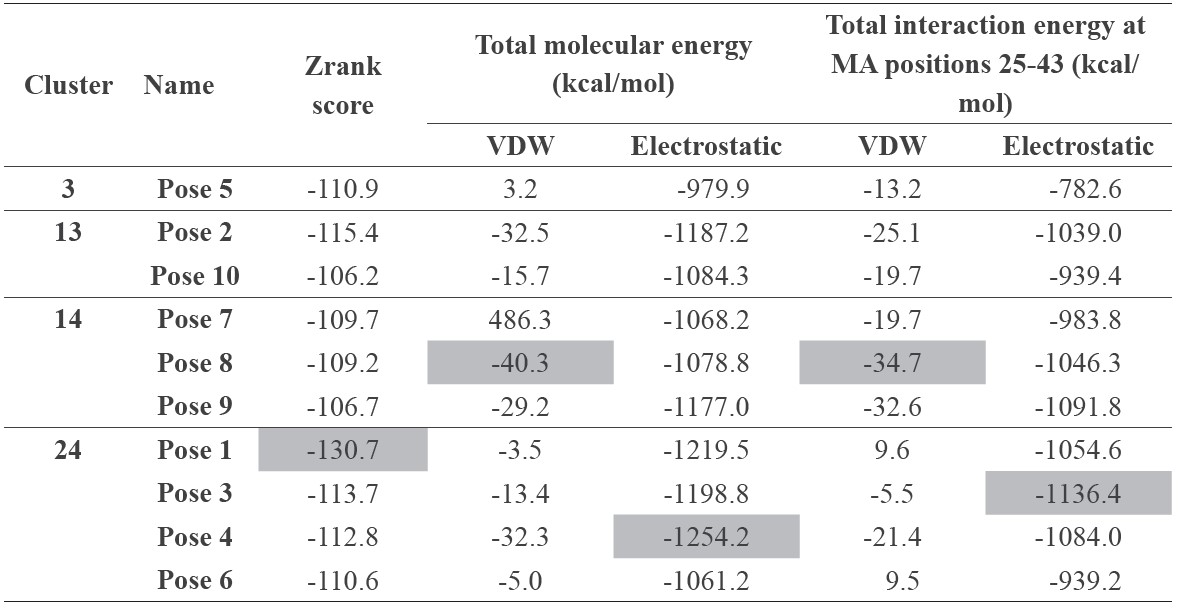
Note: The maximum value of each energy is highlighted in gray.
Prediction of binding residues involved in AnkGAGG31-MA interaction
The MA key residues interacting with AnkGAGG31 were investigated by determining the interaction energy at a single amino acid on the docking complexes (Table 3). Of the 10 docking complexes, the MA residues K27, K30, and K32 could make the intermolecular H bond to AnkGAGG31 in 7, 6, and 6 docking complexes, respectively. In terms of interaction energy, the top-five residues with the lowest interaction energy were identified in the AnkGAGG31-MA interface; the result revealed that K27, K30, and K32 still showed low interaction energy that could be found in the 9, 10, and 7 docking complexes, respectively. As shown in Figure 5, focusing on P3, which was a candidate for describing the interaction, residues K27, K30, and K32 were the top-five lowest interaction energies. For the H bond, K30 (MA:LYS30:HZ3 – AnkGAGG31: ASP100:OD1) and K32 (MA:LYS32:HZ2 – AnkGAGG31:SER67:O) promoted the stability of the AnkGAGG31-MA interaction. Moreover, the pi-cation interaction, which plays an important role in the stability of proteins, appeared in P3. The pi-cation interactions consisted of MA:K26:NZ – AnkGAGG31:P166, MA:K27:NZ – AnkGAGG31:Y36, MA:K27:NZ – AnkGAGG31:Y110, and MA:R39:NE – AnkGAGG31:Y36. Therefore, the MA residues K30, K27, and K32 were the key residues interacting with AnkGAGG31in the 10 docking poses, especially in P3. Interestingly, these residues located on the MA-helix 2 that assented to sequences of F7 and F8 (residues 25-43) from analysis of competitive ELISA.
Table 3. H bond and 5 lowest interaction energy at single MA residue interacting with AnkGAGG31 of 10 AnkGAGG31-MA docking poses.
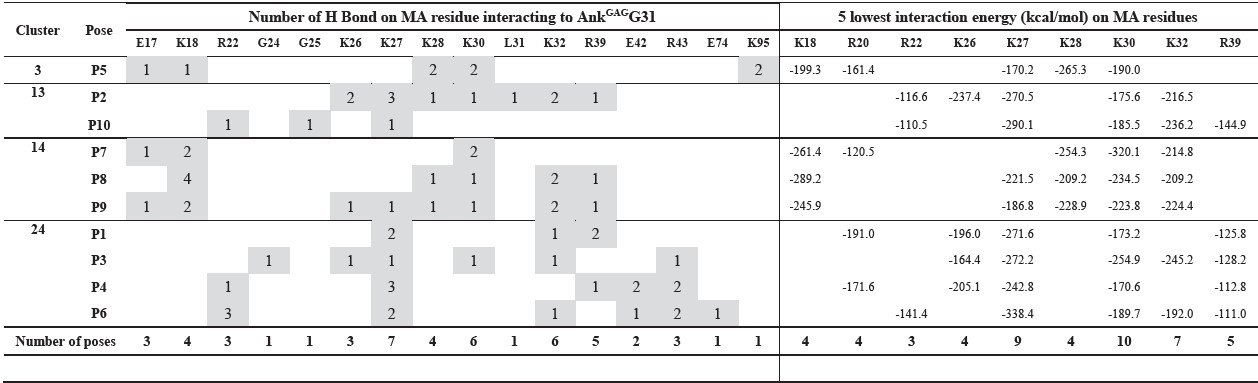
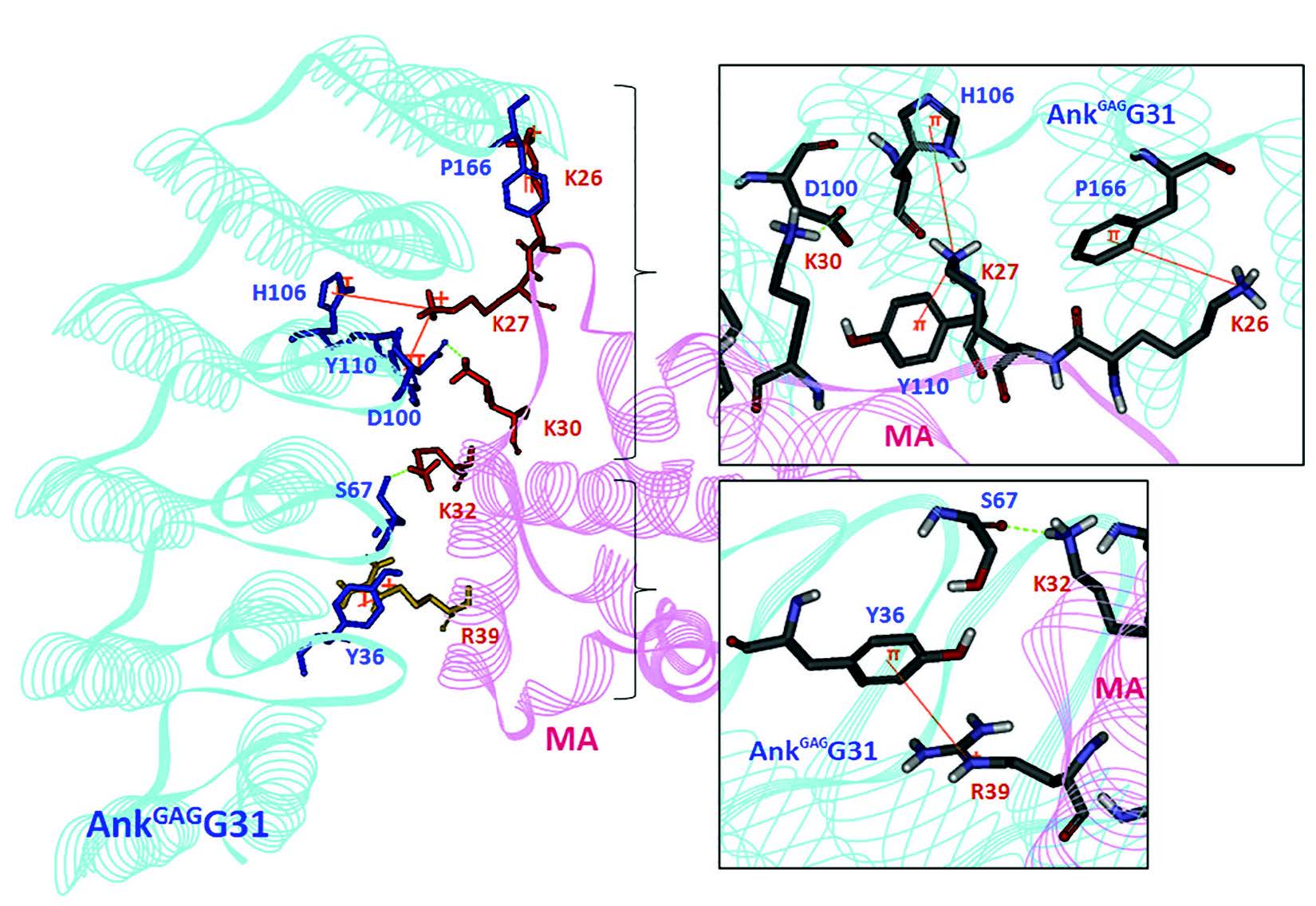
Figure 5. The interface residues between MA residues 25-43 and AnkGAGG31-MA (pose 3). The H-bond and pi-cation interactions between AnkGAGG31 (light gray) and MA (gray) are shown in gray dashed line and black thin line, respectively. The residues in the black box are presented in the different view. The H bonds occur in MA residues K30 (MA:LYS30:HZ3 – AnkGAGG31:ASP100:OD1) and K32 (MA:LYS32:HZ2 – AnkGAGG31:SER67:O). Pi-cation interactions are represented in MA residues K26 (MA:K26:NZ – AnkGAGG31:PHE166), K27 (MA:K27:NZ – AnkGAGG31:Y36 and MA:K27:NZ – AnkGAGG31:Y110), and R39 (MA:R39:NE – AnkGAGG31:Y36).
Note: Lysine (K) and arginine (R) of the interface MA residues are represented in black (or thick line, respectively.
DISCUSSION
The HIV-1 matrix is involved in many steps of the viral life cycle, including assembly and maturation (Scarlata and Carter, 2003; Bukrinskaya, 2007; Sundquist and Kräusslic, 2012; Bell and Lever, 2013). Thus, development of antibodies or alternative scaffold proteins as potential MA-inhibitors is an attractive strategy to inhibit HIV replication. In recent years, engineered ankyrin repeat proteins have been developed to more specifically bind to their targets and overcome limitations of therapeutic antibodies. Many studies have demonstrated the potential of ankyrin as a novel class of antivirals (Schweizer et al., 2008; Nangola et al., 2012; Tamaskovic et al., 2012; Khamaikawin et al., 2015; Plückthun, 2015). Scaffolds are generally used as a substitute for a biological factor, and offer great therapeutic potential. Ankyrin repeat proteins have been applied in many fields, including diagnostic and therapeutic applications. These scaffold proteins can be used as antibody alternatives in test kits for detecting the HIV protein (Nangola et al., 2014).
In terms of therapeutic applications, a number of candidates derived from these molecules are in preclinical and clinical development, with some advancing to clinical trials. Molecular Partners has developed Allergan, an ankyrin repeat protein targeting VEGF-A, that is Phase II and III clinical trials (Stahl et al., 2013, Vazquez-Lombardi et al, 2015).
In this study, we provided important information for identifying the designed scaffold protein targeting the HIV-MA protein, which could help with potential drug design. The most important criteria when developing scaffolds are to reduce the potential safety risks and provide proof of concept for future therapeutics in humans. To prove this concept, the immunogenicity and toxicity of the scaffold protein must be evaluated before applying the designed protein scaffolds to target and neutralize the HIV- MA protein. As a preliminary evaluation, we achieved the T-cell epitope prediction for AnkGAGG31 using the online tool T-Cell Epitope Prediction Tools (http://tools.iedb.org/processing/help/); we found negative major histocompatibility complex (MHC) class I and II scores compared to human ankyrin amino acid sequences. This preliminary data showed that the artificial ankyrin should perform in animal trials, although its toxicity could not be determined yet.
For preparation of the target biotinylated protein, H6-MA-BCCP was produced in vivo in Origami B harboring the pAK400cb-MA. The pAK400cB expression vector contained the BCCP gene, which is located down-stream of the inserted gene region, allowing expression of recombinant protein tagged with BCCP (Tayapiwatana et al., 2006). The C-terminal residues of BCCP were revealed to be the natural biotin binding site. This allowed for rapid and specific conformations of naturally biotinylated proteins to be produced. Furthermore, disulfide bonding and biotin conjugation can occur simultaneously under oxidizing conditions in Origami B cytoplasm (Scholle et al., 2004; Tayapiwatana et al., 2006; Orrapin et al., 2014). Therefore, in vivo non-chemical biotinylation might be more suitable than an in vitro chemical method. Furthermore, the combined screening platform using a semi-automatic machine and utilizing high-affinity streptavidin-biotinylated MA interaction was performed to efficiently select the ankyrin binder (Figure 1). The phage-displayed ankyrin library panning has been well established as a useful tool for selecting specific and high affinity binders. With the semi-automated platform, more efficient and less time-consuming phage display panning is feasible by utilizing magnetic bead nanoparticles (Konthur et al., 2010; Chin et al., 2016). This technique takes advantage of the natural high affinity between streptavidin and biotin to allow immobilization of biotinylated proteins on the streptavidin magnetic beads (Ackerman et al., 2009; Chin et al., 2016). Accordingly, one ankyrin clone with a high affinity and specificity for the MA target, referred to as AnkGAGG31, was selected (Figure 1C). This soluble tetramodular AnkGAGG31 was able to bind the MA in the conformational and truncated HIV-1 Gag polyprotein.
In the experimental study, we used two different conformational structures of MA molecule to identify the binding domain of AnkGAGG31. The biotinylated MA-fragments containing three α-helical domains were expected to be a near-native-like conformational structure, whereas the synthetic pentadecapeptides were proposed for the specific binding residues on linear epitopes. These fragments and peptides were used as competitors of the AnkGAGG31-MACA interaction. The principle of competitive ELISA was employed with the ankyrin-mediated capture of a target protein, followed by enzyme-linked immunoassay (AMELIA), which was utilized to identify the binding residues of AnkGAG1D4 on CA (referred to as ankyrinotopes) (Praditwongwan et al., 2014). The results clearly showed that AnkGAGG31 bound to MA that is located in the approximate region of residues 25-43, corresponding to the α-helix 2 (Figures 3B and 3C). According to computational studies, molecular docking revealed that AnkGAGG31 bound to MA-helix 2 in the 10 lowest energy docked models. As seen in Figure 5, this strongly supported that the MA residues 25GKKKYRLKHLVWASRELER43 on helix 2 containing highly polar residues were a good target for the electric potential on AnkGAGG31. Of ten complexes, MA residues K27, K30, and K32 importantly interacted with AnkGAGG31, since the intermolecular H bond and the top-five lowest interaction energy (vdW and electrostatics) were found on these residues. Therefore, the in vitro and in silicon experiments were in agreement that MA-helix 2, especially residues K27, K30, and K32, was responsible for the AnkGAGG31 binding interaction.
Many studies have demonstrated regions of 132 residues of MA involved in the packaging of viral RNA genome into particles, assembly, and targeting Gag multimerization to the plasma membrane (Fiorentini et al., 2006; Bukrinskaya, 2007; Sundquist and Kräusslic, 2012). The MA residues 25-43 are considered critical for proper Gag assembly and the incorporation of the envelope glycoprotein (Env) spikes during virus assembly. These assembly processes consist of: (i) the highly basic region spanning residues 18-32 that are capable of binding to the anionic heads of the lipid bilayer and electrostatically interact with phosphatidylinositol-4, 5-bisphospate of the plasma membrane, (ii) the motif residues 30-33 (30KLKH34) that are required for Env incorporation, and (iii) the sequence of 26KKK28 that is a nuclear localization signal (Fiorentini et al., 2006; Ono, 2010; Sundquist and Kräusslic, 2012; Lingappa et al., 2014). Interestingly, a similar recognition site on MA-helix 2 was also observed in small-molecule ligands (Thiadiazolanes) targeting the RNA binding site on MA (Alfadhli et al., 2013). Thiadiazolanes ability to inhibit the Gag targeting of plasma membranes has been investigated; Alfadhlin et al. (2013) proposed that this molecule binds to MA at the C-terminal portion of helix 2 (residue 40-43). Nevertheless, further studies are needed that focus on the biological role and optimization of these regions for therapeutics in clinical trials. Thus, AnkGAGG31 could probably interrupt the initial step of assembly and Env incorporation, reducing new viral production. Consequently, its antiviral effect on HIV-1 infection needs further clarification. In addition, the novel “bispecific ankyrin”, which is a fusion protein of MA-ankyrin binder (AnkGAGG31) with CA-ankyrin binder (AnkGAG1D4) (Nangola et al., 2012), could be developed as a potential inhibitor of HIV-1 multiplication. This bispecific molecule should promote the efficiency of therapeutic antiretroviral agents by inhibiting the Gag polyprotein assembly or interrupting the proteolytic cleavage machinery at the MA-CA domain. Accordingly, our results could provide useful information for further development of a new and alternative anti-HIV-1 agent that could inhibit the assembly and maturing of HIV-1 for use in HIV-treatment therapy.
CONCLUSION
Our study demonstrated the potential approach of semi-automated phage-displayed library panning using high-affinity, biotinylated, MA-streptavidin, magnetic beads to identify highly specific ankyrin scaffolds. The artificial AnkGAGG31 was successfully isolated from this method. The binding of AnkGAGG31 to the MA domain was subsequently elucidated. The data from the computational analysis was correlated to the experimental results, which suggested that the binding domain of AnkGAGG31 is on the MA-helix 2 at residues 25-43 with strong binding interaction energy. These experiments point toward development of a novel scaffold protein that would interact at the critical domain for proper Gag assembly and Env incorporation. Therefore, AnkGAGG31 could provide an alternative therapeutic molecule for treating HIV infections.
ACKNOWLEDGEMENTS
This work was supported by the Research and Researchers for Industry (RRI) Program under the Thailand Research Fund (Grant number PHD58I0044 to W.T.), the National Research University project under the Thailand’s Office of the Commission on Higher Education, Center of Biomolecular Therapy and Diagnostic, Biomedical Technology Research Center, the Faculty of Associated Medical Sciences and Graduate School of Chiang Mai University, the Ph.D. Franco-Thai scholarship program (2013) of the French Government, the 50th CMU Anniversary Ph.D. Program, and the Faculty of Pharmaceutical Sciences, Burapha University. TSL would like to acknowledge the Malaysian Ministry of Higher Education through the Higher Institution Centre of Excellence (HICoE) Grant (Grant no. 311/CIPPM/4401005) for use of its facilities. We would also like to thank the NIH AIDS Research and Reference Reagent Program for providing the pNL4-3 plasmid and a panel of overlapping MA-peptides. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 Consensus A Gag Peptides - Complete Set, Cat#8116, Lot#1. We also thank the National Electronics and Computer Technology Center (NECTC) for its support of the Discovery software.
REFERENCES
Ackerman, M., Levary, D., Tobon, G., Hackel, B., Orcutt, K.D., and Wittrup, K.D. 2009. Highly avid magnetic bead capture: an efficient selection method for de novo protein engineering utilizing yeast surface display. Biotechnology Progress. 25(3): 774-83. https://doi.org/10.1002/btpr.174
Alfadhli, A., McNett, H., Eccles, J., Tsagli, S., Noviello, C., Sloan, R., López, C.S., Peyton, D.H., and Barklis, E. 2013. Analysis of small molecule ligands targeting the HIV-1 matrix protein-RNA binding site. Journal of Biological Chemistry. 288(1): 666-76. https://doi.org/10.1074/jbc.M112.399865
Arts, E.J., and Hazuda, D.J. 2012. HIV-1 antiretroviral drug therapy. Cold Spring Harbor Perspectives in Medicine. 2(4): a007161. https://doi.org/10.1101/ cshperspect.a007161
Bell, N.M., and Lever, A.M. 2013. HIV gag polyprotein: processing and early viral particle assembly. Trends in Microbiology. 21(3): 136-44. https://doi.org/ 10.1016/ j.tim.2012.11.006
Binz, H.K., Amstlütz, P., and Plückthun, A. 2005. Engineering novel binding proteins from nonimmunoglobulin domains. Nature Biotechnology. 23(10): 1257-68. https://doi.org/10.1038/nbt1127
Binz, H.K., Strumpp, M.T., Forrer, P., and Amstlütz, A. 2003 Designing repeat protein: well-expressed, soluble and stade proteins from combinatoral libraries of consensus ankyrin repeat proteins. Journal of Molecular Biology. 333(2): 489-503. https://doi.org/10.1016/s0022-2836(03)00896-9
Bukrinskaya, A. 2007. HIV-1 matrix protein: a mysterious regulator of the viral life cycle. Virus Research. 124(1-2): 1-11. https://doi.org/10.1016 /j.virusres.2006.07.001
Chen, R., Li, L., and Weng, Z. 2003. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 52(1): 80-7. https://doi.org/10.1002/prot.10389
Chen, W.T., Shiu, C.S., Yang, J.P., Simoni, J.M., Fredriksen-Goldsen, K.I., Lee, T.S., and Zhao, H. 2013. Antiretroviral therapy (ART) side effect impacted on quality of life, and depressive symptomatology: a mixed-method study. Journal of AIDS and Clinical Research. 4: 218. https://doi.org/10.4172/2155-6113.1000218
Chin, C.F., Ler, L.W., Choong, Y.S., Ong, E.B., Ismail, A., Tye, G.J., and Lim, T.S. 2016. Application of streptavidin mass spectrometric immunoassay tips for immunoaffinity based antibody phage display panning. Journal of Microbiological Methods. 120: 6-14. https://doi.org/10.1016/j.mimet.2015.11.007
Fiorentini, S., Marini, E., Caracciolo, S., and Caruso, A. 2006. Functions of the HIV-1 matrix protein p17. New Microbiologica. 29(1): 1-10.
Fiorentini, S., Marsico, S., Becker, P.D., Iaria, M.L., Bruno, R., Guzmánc, C.A., and Carusoa, A. 2008. Synthetic peptide AT20 coupled to KLH elicits antibodies against a conserved conformational epitope from a major functional area of the HIV-1 matrix protein p17. Vaccine. 26(36): 4758-65. https://doi.org/ 10.1016/j.vaccine.2008.06.082
Gilbreth, R.N., and Koide, S. 2012. Structural insights for engineering biding proteins based on non-antibody scaffolds. Current Opinion in Structural Bilogy. 22(4): 413-420. https://doi.org/10.1016/j.sbi. 2012.06.001
Halperin, I., Ma, B., Wolfson, H., and Nussinov, R. 2002. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins. 47(4): 409-43. https://doi.org/10.1002/prot.10115
Khamaikawin, W., Saoin, S., Nangola, S., Chupradit, K., Sakkhachornphop, S., Hadpech, S., Onlamoon, N., Ansari, A.A., Byrareddy, S.N., Boulanger, P., et al. 2015. Combined antiviral therapy using designed molecular scaffolds targeting two distinct viral functions, HIV-1 genome integration and capsid assembly. Molecular Therapy —Nucleic Acids. 4: e249. https://doi.org/10.1038/mtna.2015.22
Konthur, Z., Wilde, J., and Lim, T. S. 2010. Semi-automated magnetic bead-based antibody selection from phage display libraries. In: Kontermann, R., and Dübel, S. (eds) Antibody Engineering. Springer Berlin Heidelberg, Berlin. p. 267–287.
Li, L., Chen, R., and Weng, Z. 2002. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 53(3): 693-707. https://doi.org/10.1002/prot.10460
Lingappa, J.R., Reed, J.C., Tanaka, M., Chutiraka, K., and Robinson, B.A. 2014. How HIV-1 Gag assembles in cells: Putting together pieces of the puzzle. Virus Research. 193: 89-107. https://doi.org/10.1016/j.virusres.2014.07.001
Luque, F., Oya, R., Macias, D., and Saniger, L. 2005. Gene therapy for HIV-1 infection: are lethal genes a valuable tool. Cellular and Molecular Biology (Noisy-le-Grand, France). 51(1): 93-101. https://doi.org/10.1170/T601
Muriaux, D., Darlix, J.L., and Cimarelli, A. 2004. Targeting the assembly of the human immunodeficiency virus type I. Current Pharmaceutical Design. 10(30): 3725-39. https://doi.org/10.2174/1381612043382701
Nangola, S., Thongkum, W., Saoin, S., Ansari, A.A., and Tayapiwatana, C. 2014. An application of capsid-specific artificial ankyrin repeat proteinproduced in E. coli for immunochromatographic assay as a surrogate for antibody. Applied Microbiology and Biotechnology. 98(13): 6095-103. https://doi.org/10.1007/s00253-014-5777-5
Nangola, S., Urvoas, A., Valerio-Lepiniec, M., Khamaikawin, W., Sakkhachornphop, S., Hong, S.S., Boulanger, P., Minard, P., and Tayapiwatana, C. 2012. Antiviral activity of recombinant ankyrin targeted to the capsid domain of HIV-1 Gag polyprotein. Retrovirology. 9: 17. https://doi.org/10.1186/1742-4690-9-17
Ono, A. 2010. Relationships between plasma membrane microdomains and HIV-1 assembly. Biology of the Cell. 102(6): 335-50. https://doi.org/ 10.1042/BC20090165
Orrapin, S., and Intorasoot, S. 2014. Recombinant expression of novel protegrin-1 dimer and LL-37-linker-histatin-5 hybrid peptide mediated biotin carboxyl carrier protein fusion partner. Protein Expression and Purification. 93: 46-53. https://doi.org/10.1016/j.pep.2013.10.010
Paredes, R., and Clotet, B. 2010. Clinical management of HIV-1 resistance. Antiviral Research. 85(1): 245-65. https://doi.org/10.1016/j.antiviral.2009.09.015
Pierce, B., and Weng, Z. 2008. A combination of rescoring and refinement significantly improves protein docking performance. Proteins. 72(1): 270-9. https://doi.org/10.1002/prot.21920
Plückthun, A. 2015. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annual Review of Pharmacology and Toxicology. 55: 489-511. https://doi.org/10.1146/annurev-pharmtox-010611-134654
Praditwongwan, W., Chuankhayan, P., Saoin, S., Wisitponchai, T., Lee, V.S., Nangola, S., Hong, S.S., Minard, P., Boulanger, P., Chen, C.J., et al. 2014. Crystal structure of an antiviral ankyrin targeting the HIV-1 capsid and molecular modeling of the ankyrincapsid complex. Journal of Computer-Aided Molecular Design. 28(8): 869-84. https://doi.org/10.1007/s10822-014-9772-9
Reddy, R.B., Reddy, P.L., and Saragandla, S. 2012. Structure-guided modeling and binding studies of GABAA receptor subunit beta-3. International Journal of Computer Applications. 15(59): 42-6. https://doi.org/0.5120/9627-4275
Saoin, S., Wisitponchai, T., Intachai, K., Chupradit, K., Moonmuang, S., Nangola, S., Kitidee, K., Fanhchaksai, K., Lee, V.S., Hong, S.S., et al. 2017. Deciphering critical amino acid residues to modify and enhance the binding affinity of ankyrin scaffold specific to capsid protein of human immunodeficiency virus type 1. Asian Pacific Journal of Allergy and Immunology. 36(2): 126-135. https://doi.org/10.12932/AP-280217-0037
Sawyer, N., Speltz, E.B., and Regan, L. 2013. NextGen protein design. Biochemical Society Transactions. 41(5): 1131-6. https://doi.org/10.1042/BST20130112
Scarlata, S., and Carter, C. 2003. Role of HIV-1 gag domains in viral assembly. Biochimica et Biophysica Acta. 1614(1): 62-72. https://doi.org/10.1016/S0005-2736(03)00163-9
Scholle, M.D., Collart, F.R., and Kay, B.K. 2004. In vivo biotinylated proteins as targets for phage-display selection experiments. Protein Expression and Purification. 37(1): 243-52. https://doi.org/10.1016/j.pep.2004.05.012
Schweizer, A., Rusert, P., Berlinger, L., Ruprecht, C.R., Mann, A., Corthésy, S., Turville, S.G., Aravantinou, M., Fischer, M., Robbiani, M., et al. 2008. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLOS Pathogens. 4(7): e1000109. https://doi.org/ 10.1371/journal.ppat.1000109
Sticht, J., Humbert, M., Findlow, S., Bodem, J., Muller, B., Dietrich, U., Werner, J., and Kräusslich, H.G. 2005. A peptide inhibitor of HIV-1 assembly in vitro. Nature Structural & Molecular Biology. 12(8): 671-7. https://doi.org/10.1038/nsmb964
Stahl, A., Stumpp, M.T., Schlegel, A., Ekawardhani, S., Lehrling, C., Martin, G., Gulotti-Georgieva, M., Villemagne, D., Forrer, P., Agostini, H.T., et al. 2013. Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis. 16(1): 101-111. https://doi.org/ 10.1007/s10456-012-9302-0
Sundquist, W.I., and Kräusslich, H.G. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harbor Perspectives in Medicine. 2(7): a006924. https://doi.org/10.1101/cshperspect.a006924
Tamaskovic, R., Simon, M., Stefan, N., Schwill, M., and Plückthun, A. 2012. Designed ankyrin repeat proteins (DARPins) from research to therapy. Methods in Enzymology. 503: 101-34. https://doi.org/10.1016/B978-0-12-396962-0.00005-7
Tayapiwatana, C., Chotpadiwetkul, R., and Kasinrerk, W. 2006. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. Journal of Immunological Methods. 317(1-2): 1-11. https://doi.org/10.1016/j.jim.2006.07.024
Tue-ngeun, P., Kodchakorn, K., Nimmanpipug, P., Lawan, N., Nangola, S., Tayapiwatana, C., Rahman, N.A., Zain, S.M., and Lee, V.S. 2013. Improved scFv anti-HIV-1 p17 binding affinity guided from the theoretical calculation of pairwise decomposition energies and computational alanine scanning. BioMed Research International. 731585. https://doi.org/10.1155/2013/713585
Vazquez-Lombardi, R., Phan, T.G., Zimmermann, C., Lowe, D., Jermutus, L., and Christ, D. 2015. Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today. 20(10): 1271-1283. https://doi.org/10.1016/j.drudis.2015.09.004
Vyas, V.K., Ukawala, R.D., Ghate, M., and Chintha, C. 2012. Homology modeling a fast tool for drug discovery: current perspectives. Indian Journal of Pharmaceutical Sciences. 74(1): 1-17. https://doi.org/10.4103/0250-474X.102537
Wiehe, K., Peterson, M.W., Pierce, B., Mintseris, J., and Weng, Z. 2008. Protein-protein docking: overview and performance analysis. Methods in Molecular Biology. 413: 283-314. https://doi.org/10.1007/978-1-59745-574-9_11
Wisitponchai, T., Nimmanpipug, P., Lee, K.W., and Tayapiwatana, C. 2013. MATLAB process for validating amino acids on CD4 involving in DARPin binding site from ZDOCK molecular docking Database. Chiang Mai Journal of Science. 40(2): 233-39.
Wisitponchai, T., Shoombuatong, W., Lee, V.S., Kitidee, K., and Tayapiwatana, C. 2017. AnkPlex: algorithmic structure for refinement of near-native ankyrin-protein docking. BMC Bioinformatics. 18(1): 220. https://doi.org/10.1186/s12859-017-1628-6
Weeraya Thongkum1,2, Kanokwan Samerjai1,2, Somphot Saoin2,3, Tanchanok Wisitponchai2, Sudarat Hadpech1,2, Vannajan Sanghiran Lee4, Theam Soon Lim5, and Chatchai Tayapiwatana1,2*
1 Division of Clinical Immunology, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand
2 Center of Biomolecular Therapy and Diagnostic, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand
3 Division of Clinical Immunology and Transfusion Sciences, School of Allied Health Sciences, University of Phayao, Phayao 56000, Thailand
4 Department of Chemistry, Faculty of Science, University of Malaysia, Kuala Lumpur 50603, Malaysia
5 Institute for Research in Molecular Medicine, Universiti Sains Malaysia, Penang 11800, Malaysia
*Corresponding author. E-mail: asimi002@hotmail.com
Total Article Views
Article history:
Received: December 6, 2017
Revised: March 23, 2018
Accepted: April 19, 2018