 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

The Vascular Endothelial Growth Factor Pathway of Angiogenesis in Tumors: Associated Pharmaceutical Targets and Treatments
David Patrick MaisonPublished Date : 2018-10-01
DOI : https://doi.org/10.12982/CMUJNS.2018.0023
Journal Issues : Number 4 , October-December 2018
ABSTRACT
Tumor angiogenesis is a cellular and molecular process in many species that is responsible for the sprouting and development of blood vessels into a tumor. This vasculature supplies a tumor with nutrients and oxygen. This supply is an absolute requirement for solid tumors to grow and become metastatic. The pathway associated with VEGF (Vascular Endothelial Growth Factor) ligands and VEGF receptors is considered the primary pathway of the tumor angiogenesis process. This review first outlines the VEGF-pathway of tumor angiogenesis, focusing on the VEGF ligands and receptor cascades resulting in delta-like-ligand 4 (DLL4) and downstream intercellular reactions with Notch. The review commences at a tumor's oncogenic switch to the angiogenic phenotype, and concludes at the completion of angiogenesis – the establishment of functional tumor vasculature and enhanced metastatic capabilities. Second, this review provides an overview of current pharmaceutical tumor treatments exclusively targeting the VEGF pathway of angiogenesis, including a basic summary of the primary VEGF pathway-targeted drugs, with a focus on drug targets and Food & Drug Administration (FDA) approval status for indicated forms of cancer. Finally, this review discusses novel and hypothetical mechanisms to target tumor angiogenesis with therapeutics, focusing on two established targeting devices and proposing one possible mechanism utilizing the complement system that targets vasculature in a manner mimicking type II hypersensitivity with chimeric complement.
Keywords: Cancer, Angiogenesis, Antibodies, Cancer therapy, VEGF
INTRODUCTION
Cancer is a dominant cause of mortality among humans, responsible for more than eight million deaths in 2015 (World Health Organization, 2018). Together, cancers encompass a group of diseases characterized by abnormal cellular growths known as tumors. Under certain conditions, these growths can invade and infiltrate almost any part of the human body. Often arising within a single normal cell, genetic alterations cause that cell to change extracellular expressions and exocytosis outputs to that of the angiogenic phenotype – a phenotype term directly referencing the expression of growth factors correlating to vascular endothelial cell recruitment (El-Kenawi and El-Remessy, 2013). The genetic alterations that cause a normal cell to switch to become a cancer cell also change the expression of factors that induce and stimulate angiogenesis. Angiogenesis is an intricate, coordinated procession of both molecular and cellular events through which new blood vessels sprout from the existing systemic vasculature (Ferrara and Adamis, 2016). When a tumor can produce sufficient proangiogenic factors to induce angiogenesis to the point of complete vascularization, then the primary tumor can grow and become metastatic. Therefore, angiogenesis is the limiting step for a dormant cancer cell to metastasize (Folkman, 1971, 2006, 2007).
Since the discovery that angiogenesis serves a rate-limiting role in the establishment of a tumor (Folkman, 1971), progression in anti-angiogenesis drug development has been significant, with greater access, availability, and variety. Notable progress includes therapeutics that antagonize or inhibit synthesis of pro-angiogenic factors and therapeutics that target receptors on vascular endothelial cells for those factors (Folkman, 2003). A significant advance in angiogenesis inhibitor treatment was the development of monoclonal antibodies directed to VEGF (Ferrara et al., 2004, 2005). Monoclonal antibodies can bind specifically to almost any molecular or biochemical sequence, and generally, serve in an antagonistic role to block a particular protein. In the past decade, increases in knowledge of the molecular processes of the VEGF angiogenesis pathway point to possible alternative angiogenesis inhibitor targets in tumor therapy, including DLL4 (Delta-Like Ligand 4), the Notch receptor and domains, and the VEGFR-1 receptor (Ridgway et al., 2006; Liu et al., 2014).
This review evaluates the potential of angiogenesis inhibitor therapy in tumor treatment. This review has three goals: to provide an overview of tumor angiogenesis, provide a perspective of current major angiogenesis inhibitor drug treatments, and discuss pharmacological routes of targeting the DLL4-Notch interaction within the VEGF pathway. To elaborate on the third goal, consideration of tumor angiogenesis together with novel approaches in antibody engineering reveals an alternative to the antagonistic antibody blockade and a potential alternative target within the VEGF pathway. The alternative to the antagonistic antibody blockade is antibody mediation of cytotoxicity to growing vasculature via the innate plasma complement cascade. The alternative target within the VEGF pathway is the cell-surface molecular interactions between tip-cells and stalk-cells.
TUMOR ANGIOGENESIS
Tumors
Cells with the ability to cause cancer - often identified clinically by the tumors they produce - are present in a dormant state in most humans (Folkman and Kalluri, 2004), with incidence appearing to increase with age (Kareva, 2016). When the cells comprising a tumor undergo sufficient oncogenic mutations, several significant events occur to sustain unlimited growth and immortality. Tumor immortality and unrestrained growth beyond a limited size depend upon the continuous provision of resources; thus, resource procurement is one of the critical early events in a newly active tumor. Resource procurement depends on recruitment, migration, and maturation of additional blood vessels (Folkman and Shing, 1992). As such, tumor angiogenesis is a complex, coordinated, and intricate process that is gradually achieved via many signaling cascades secreted from the tumor, and is the tumors mechanism of resource procurement. The signaling pathway of focus here is the VEGF pathway.
The tumor first secretes enzymes – such as matrix metalloproteinase (Folkman, 2007) – to break down the surrounding connective tissue. The tumor simultaneously secretes proangiogenic factors into the voided areas to form pro-angiogenic chemical “gradients of growth factors and chemokines that attract nascent vessels” (R. Cohen, personal communication). When a pro-angiogenic gradient encounters a blood vessel, enzymes detach the mural cells that envelope vascular endothelial cells, degrade the basement membrane, and caue exposure of luminal vascular endothelial cells (Kandel et al., 2011) (Figure 1). When the signaling gradient then encounters the exposed vascular luminal endothelial cells, these cells undergo morphological changes that allow them to follow the path of the pro-angiogenic gradient. These initial angiogenic changes and events precede tip cell selection, vascular endothelial cell sprouting, migration, morphogenesis, and vascular maturation (Kandel et al., 2011). The steps involved in the VEGF gradient are among the primary targets of current cancer therapeutics.
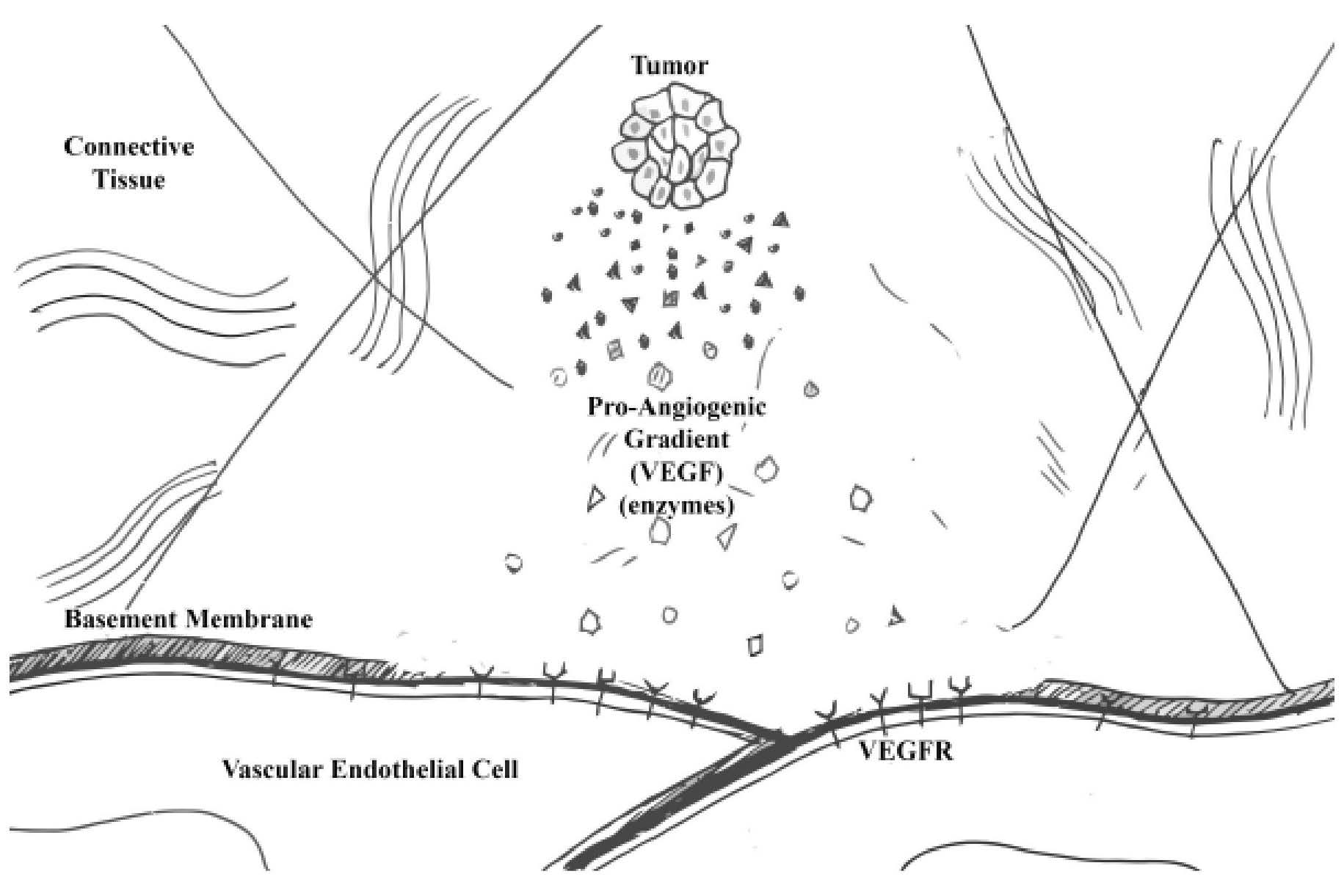
Figure 1. Introduction to tumor angiogenesis.
Tumor cells expressing the angiogenic phenotype are those that are actively recruiting blood vessels. Tumor cells actively recruiting blood vessels use enzymes to degrade the surrounding connective tissue. Along with the enzymes, the tumor cells also exocytose vascular endothelial growth factors (VEGF). When the pro-angiogenic gradient encounters a vascular endothelial cell, degradative proteins remove the basement membrane and expose the vascular endothelial growth factor receptors (VEGFR) to VEGF.
Primary tumor angiogenic gradient constituents
Among the chemicals secreted by an immortal tumor are pro-angiogenic factors including the ligands of the VEGF family (VEGF-A, -B, -C, -D, and -E) and placenta growth factor (PGF) (Holmes et al., 2007; Ferrara and Adamis, 2016). These ligands variably serve as agonists for the three forms of VEGFR (VEGFR-1, -2, -3) (Ferrara and Adamis, 2016). Holmes et al. (2007) describe the protein components of the primary angiogenic gradient. Table 1 summarizes these primary components.
Table 1. Vascular endothelial growth factor receptors.
When a blood vessel is enzymatically relieved of its protective coating and basement membrane by a tumor’s pro-angiogenic gradient, then the underlying vascular endothelial cells are exposed to varying concentrations of various forms and isoforms of VEGF ligands, which stimulates angiogenesis. Vascular endothelial cells express VEGFR-2 and -3 on the non-luminal/basolateral membrane. The binding of VEGF-A from a tumor to VEGFR-2 and -3 on these vascular endothelial cells triggers a process known as tip cell selection (Boareto et al., 2015).
Tip cell selection
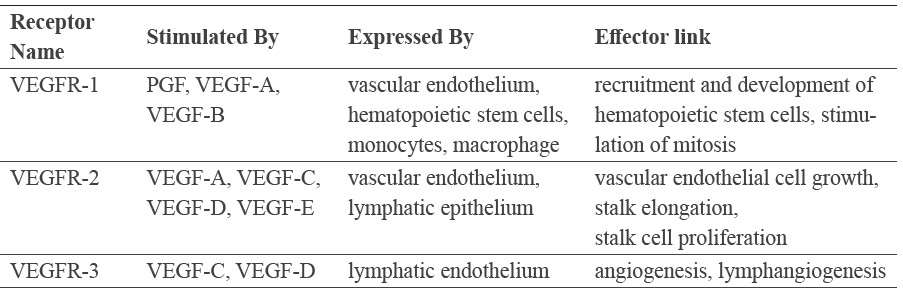
Boareto et al. (2015) describe the following molecular principles of tip cell selection. Tip cell selection, also known as lateral inhibition, is a race among vascular cells to molecularly self-select and un-select neighbors (Figure 2). This race begins whenever a tumor’s proangiogenic gradient encounters quiescent vascular endothelium. Vascular endothelial cells take on individual qualities when competing for selection as a tip-cell – and thereby first prevent neighboring cells from self-selecting as tip-cells, and instead, metaphorically tether their neighbors in servitude as stalk cells. Adjacent cells to tip-cells, therefore, molecularly and morphologically alter to become stalk cells. Stalk cells and tip cells are thus functionally different in role, phenotypically distinct in receptor expression, and morphologically different in angiogenic fate (De Smet et al., 2009).
To continue, VEGF-A isoforms binding to VEGFR-2 receptors on dormant vascular endothelial cells triggers an intracellular cascade in the vascular endothelial cells that increase surface expression of DLL4 and Jagged (Boareto et al., 2015). The DLL4 expression is the first characteristic of a newly selected tip cell (Patel et al., 2005; De Smet et al., 2009). Jagged participates in a feedback loop to further commit tip cells to their fate by reducing expression of VEGFR-1, as well as functioning intercellular to increase vessel stability (Boareto et al., 2015). DLL4 binding to Notch receptors on adjacent cells confines the adjacent cell to a stalk cell fate. Within the adjoining cell, ADAM (a disintegrin and metalloprotease) cleaves the NICD (Notch intracellular domain) from NECD (Notch extracellular domain) bound to DLL4 (Quillard and Charreau, 2013). NICD signals to the nucleus to increase surface expression of VEGFR-1, as well as increase exocytosis of soluble VEGFR-1 (sVEGFR-1) – assisting in further commitment and recruitment of stalk cells.
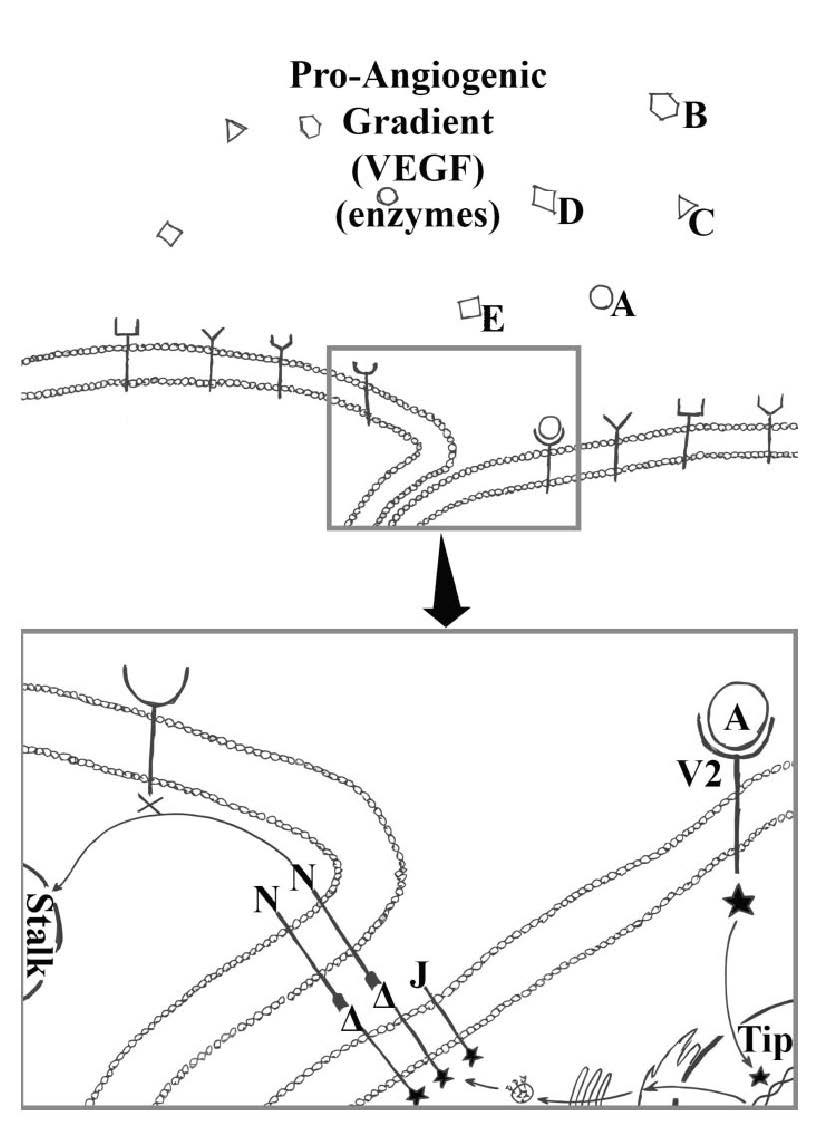
Figure 2. Tip-cell selection.
Tip-cell selection begins after basement membrane removal from vascular endothelial cells, and there is exposure of vascular endothelial cells to the pro-angiogenic gradient; in particular, the vascular endothelial growth factors (A, B, C, D, and E). The exposed endothelial basolateral surface present vascular endothelial growth factor tyrosine-kinase receptors able to bind vascular endothelial growth factors within the pro-angiogenic gradient. Vascular endothelial growth factor A (A) binding to vascular endothelial growth factor receptor 2 (V2) cause a cascade of intracellular effects resulting in a cell becoming a tip-cell ( ). A chosen tip-cell (
). A chosen tip-cell ( ) will begin to express delta-like-ligand-4 (DLL4)(Δ) and Jagged (J). DLL4 bind to and activate the Notch receptors (N) on adjacent vascular endothelial cells. Activation of the Notch causes a cascade of intracellular effects causing the adjoining cell to become a stalk cell. Jagged further commit tip cells to their fate as well as function in vessel stability.
) will begin to express delta-like-ligand-4 (DLL4)(Δ) and Jagged (J). DLL4 bind to and activate the Notch receptors (N) on adjacent vascular endothelial cells. Activation of the Notch causes a cascade of intracellular effects causing the adjoining cell to become a stalk cell. Jagged further commit tip cells to their fate as well as function in vessel stability.
As a review to our current progression, an immortal tumor secretes VEGF that binds to VEGFR-2 on a random vascular endothelial cell. VEGFR-2 binding causes an increased expression of DLL4 and Jagged that essentially commits that cell to a tip cell fate. DLL4 binding to Notch on neighboring cells increases the neighboring (stalk) cells expression of VEGFR-1, which is the selection of stalk cells. Following the assignment of cellular fates via the selection process, the migration and elongation process ensues.
Migration and elongation
Migration and elongation is a guided growth process that begins with morphological changes to a vascular endothelial cell selected as a tip cell. These morphological changes, introduced by way of exposure to VEGF, start with the development of a transverse arc actin meshwork in the direction of migration (Mayor and Etienne-Manneville, 2016). This meshwork allows the tip cell to project filopodia (actin filament bundles), which probe the stromal environment for the highest concentration of pro-angiogenic factors (Gerhardt et al., 2003), giving dimensional directionality to growth. Upon coordination of migration direction, a connected intracellular-intercellular cascade of signals transfer through the non-proliferative tip cell to the proliferative stalk-cells, elongating the sprout as the stalk cells divide (Figure 3).
Investigators have proposed two hypotheses of vesicular elongation, with a third supposed from their combination. Leung et al. (1989) proposed that VEGF serves as a vascular endothelial cell mitogen; meaning that, via linked intracellular and intercellular cascades, VEGF binding to VEGFR-2 on a tip cell causes adjacent stalk cells to commence mitosis and propel the tip cell in its gradient orientated direction. In a competing hypothesis, sprouts elongate by a rolling adhesion insertion mechanism, which features incorporation of hematopoietic stem cells and endothelial progenitor cells, each expressing VEGFR-1, -2, and each secreting soluble VEGFR-1 (Folkman, 2007; Melero-Martin and Dudley, 2011). This second hypothesis additionally postulates that endothelial progenitor cells are the exclusive cells that form the lumen of angiogenic vessels (Melero-Martin and Dudley, 2011). A third alternative hypothesis supposes from both elongation processes. Meaning that tumor angiogenesis may utilize both proposed elongation processes – determined by the availability of endothelial progenitor cells, the availability of mitogens, or the progression of stalk elongation. Regardless of the exact mechanism of elongation, however, a wide acceptance is that elongation commences from the base of the sprout and filopodia projections guide the tip cell (Gerhardt et al., 2003; De Smet et al., 2009).
Figure 3. Migration and elongation.
(A) Migration begins with a selected tip cell forming a transverse arc actin meshwork to project filopodia – similar to the way microvilli project in the gut epithelium. The tip-cell filopodia express vascular endothelial growth factor receptor 2 (VEGFR2), which bind to vascular endothelial growth factor A (VEGFA) within the pro-angiogenic gradient and trigger intercellular cascades to adjust the actin microfilaments accordingly. Vascular endothelial cells selected as stalk cells secrete soluble vascular endothelial growth receptors (sVEGFR) to stimulate adjacent vascular endothelial cells further to become stalk cells as well as prevent the neighboring cells from becoming tip-cells via binding to VEGF.
(B) Elongation begins with morphological changes to angiogenic cells. Intercellular interactions between the top tip cell and the juxtaposed stalk cells result in morphological changes. These morphological changes give stability and allow the vessel to grow in a particular direction.
Morphogenesis and completion
For clarity, we first review up to the current position in the progression of tumor angiogenesis. A dormant tumor cell underwent genetic alterations (oncogenic mutations) sufficient to express the angiogenic phenotype and become an immortal tumor. An immortal tumor first requires nutrients, so the tumor degraded its surrounding environment, and simultaneously, the tumor sent out gradients of enzymes and pro-angiogenic ligands (VEGF) in search of blood vessels. Once a gradient located a blood vessel, the blood vessel was relieved of its protective barriers by tumor enzymes to expose quiescent vascular endothelial cells. The dormant vascular endothelial cells express VEGFR-2, which, when bound by VEGF, causes those endothelial cells to compete for tip cell selection. Selected tip cells orient in the direction of the pro-angiogenic gradient via apical projection of filopodia and non-apical expression of DLL4. Expressed DLL4 binds to Notch on adjacent cells, causing an intracellular cascade that induces expression of VEGFR-1 and Jagged on the neighboring cell. Continual binding of VEGF ligands to VEGFR on the tip cell causes an intercellular cascade along the sprout that triggers the base stalk cells to proliferate, or progenitor cells to incorporate, thus elongating the sprout.
Multiple nascent vessels must now be included in this description to provide a more comprehensive conceptual framework, as the cascade that an immortal tumor creates select many tip cells and recruit many sprouts. For pharmacological treatment, scientists must address and recognize the predicament that the many nascent sprouts are at many different phases of angiogenesis. Tip cells from different sprouts need to converge (Figure 4), undergo further morphogenesis, and form a loosely working lumen before tumor angiogenesis can be complete. Once morphogenesis establishes an initial circuit flow, vasculature throughout the tumor remains immature, with incomplete fenestrated intercellular adhesions, chaotic organization, and compression from proliferating tumor cells in juxtaposition (Jain, 2003; El-Kenawi and El-Remessy, 2013). Thus, the vascular lumen is exposed directly to tumor cells in some places (Jain, 2003). Proliferating tumor cells, therefore, make up and maintain the luminal wall of the vasculature throughout some parts of a tumor (Jain, 2003). This maintenance of leaky and immature vessels, merely via the compression resulting from proliferating tumor cells and continual secretion of VEGF ligands, allows for rapid fusion by still more incoming sprouts. The establishment of these leaky and immature vessels gives tumors nutrients and oxygen, “enabling full expression of uncontrolled cellular proliferation and distant metastasis as tumor cells enter the vascular lumen” (R. Cohen, personal communication). Therefore, completion of angiogenesis allows an immortal tumor to grow large, metastasize, and spread to any tissue connected to the systemic vasculature.
“Tumor angiogenesis, once established, is subject to continuous remodeling as the tumor grows and evolves” (R. Cohen, personal communication). Sprouts continue to invade the tumor, and the tumor continues to metastasize and degrade the surrounding connective tissues. Whether or not vasculature outside of the tumor (the sprout: from the vessel to the tumor) remodels and matures to form complete vessels is unknown. This vessel maturation includes creating non-leaky endothelial cell junctions, developing a basement membrane, and recruiting a pericyte or other mural cell coating (Jain, 2003). Vessel maturation is typical for growing angiogenic sprouts during natural events, such as wound healing and embryonic development (Folkman and Shing, 1992; Jain, 2003; Folkman, 2006, 2007; Tahergorabi and Khazaei, 2012). However, the immune system tightly regulates the angiogenic extracellular environment during wound healing and embryonic development; a property not stringently paralleled in tumor angiogenesis. Presumably, the particular tumor microenvironment dictates the extent of the maturation process, although experts note that tumor vasculature is structurally defective and may lack a pericyte cover (Ferrara and Adamis, 2016). Immune cells probably assist the tumor in turning its sprouts into vessels (metaphorically turning dirt roads into superhighways), while also biologically terraforming the surrounding landscape to accept the tumor as a newly formed entity of the body (Lodish et al., 2007).
This complex tumor angiogenesis process is currently a pharmacological target for novel tumor treatments. These treatments are collectively known as angiogenesis inhibitors. “Given the complexity of the process, pharmacologists designing and discovering angiogenesis inhibitors have a vast array of potential targets to choose from” (R. Cohen, personal communication). This extensive collection includes molecular entities of the VEGF pathway of tumor angiogenesis as possible targets, including the VEGF ligand family, the VEGFR family, DLL4, Notch, NECD, and NICD. However, targeting is often limited in efficacy due to factors including tissue expression, half-life, selectivity, and tumor vasculature permeability (R. Cohen, personal communication).
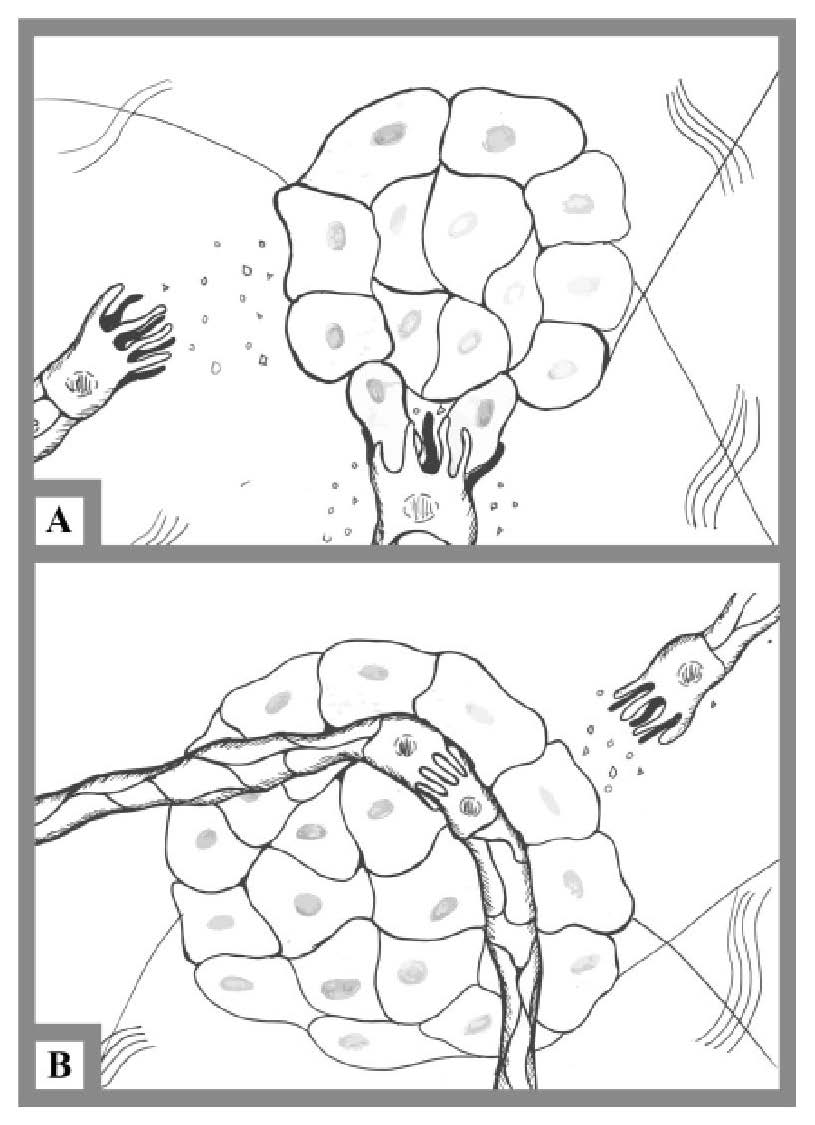
Figure 4. Morphogenesis and completion
(A) Morphogenesis involves many sprouts converging through a mass of tumor cells. Tip-cells collide with the tumor mass cells and begin to weave through the tumor. Tip-cells continue to follow the pro-angiogenic gradient through the mass of tumor cells until two or more different tip-cells converge with one another and form loosely functional vasculature.
(B) Completion involves the establishment of a weakly functional vasculature. Angiogenesis completion marks the first creation of vasculature, but the tumor continues to recruit more tip-cells – with tethered sprouts – to increase nutrient and oxygen intake. Once a tumor mass obtains vasculature, it continues to grow and metastatically invade the host.
ANGIOGENESIS INHIBITORS – VEGF-PATHWAY TARGETED THERAPY
Inhibitors of angiogenesis fit into two classes (Folkman, 2003): direct inhibitors, which are endogenous inhibitors targeting the vasculature and endothelial cells, and indirect inhibitors, which are exogenous and target activators of angiogenesis (El-Kenawi and El-Remessy, 2013). Direct inhibitors occur naturally (Folkman and Kalluri, 2004; Folkman, 2007) and often release from connective tissue resident cells when tumor secretions of proteases and matrix metalloproteinase lyse the resident cells. Indirect inhibitors block pro-angiogenic ligands, pro-angiogenic ligand receptors, or other products of tumor cell oncogenes (Folkman, 2003).
Indirect VEGF-pathway angiogenesis inhibitor therapies classify into mono-therapy and combination therapy (El-Kenawi and El-Remessy, 2013). Mono-therapy uses a single drug or treatment; combination therapy uses multiple medications or treatments, often simultaneously to provide an enhanced therapeutic effect. The current strategies to target angiogenesis in an inhibitory manner include both mono-therapy and combination therapies that principally target the VEGF-pathway (Fang and Salvan, 2011; de Lartigue, 2016).
The VEGF-pathway angiogenesis inhibitors discussed here are all National Cancer Institute (NCI) recognized, FDA approved, and indirect pharmaceutical therapies; these include axitinib, bevacizumab, cabozantinib, lenvatinib, midostaurin, pazopanib, ponatinib, ramucirumab, regorafenib, sorafenib, vandetanib, and ziv-aflibercept (Barbie and Frank, 2012; El-Kenawi and El-Remessy, 2013; de Lartigue, 2016; Food and Drug Administration, 2017; National Cancer Institute, 2018; Wishart et al., 2018). Many of these VEGF-pathway targeting drugs, although promising in molecular design and reasoning, have proven less promising for broad clinical application, and thus require further research.
Barbie and Frank (2012), Welti et al. (2013), El-Kenawi and El-Remessy (2013), de Lartigue (2016), Ferrara and Adamis (2016), Food and Drug Administration (2017), National Cancer Institute (2018), Wishart et al. (2018) all describe the available VEGF-pathway pharmacological treatments. Table 2 summarizes these treatments.
Table 2. Drugs targeting the VEGF pathway of angiogenesis in tumor therapy.
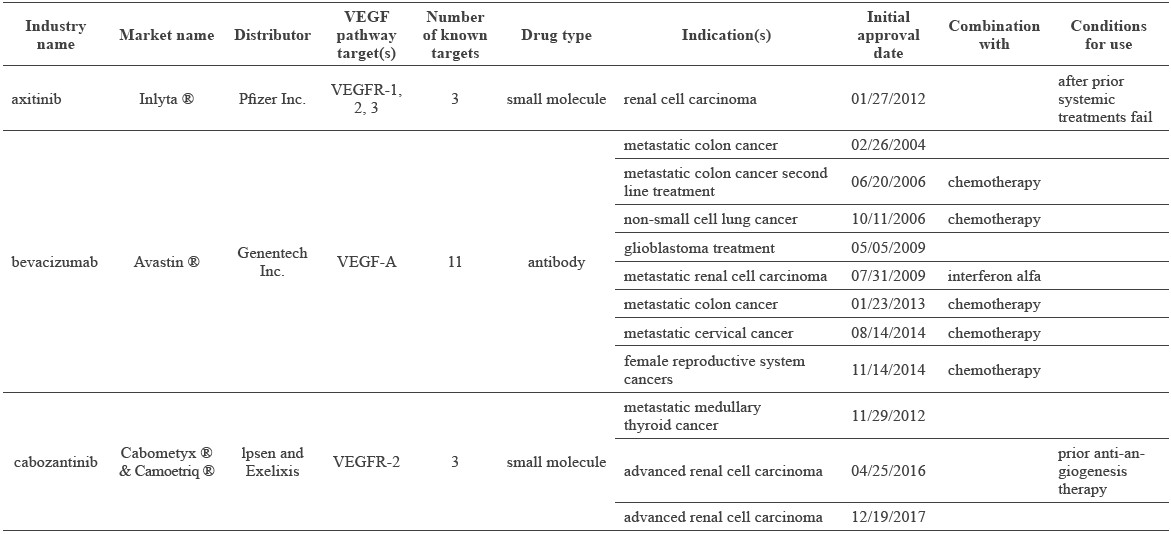
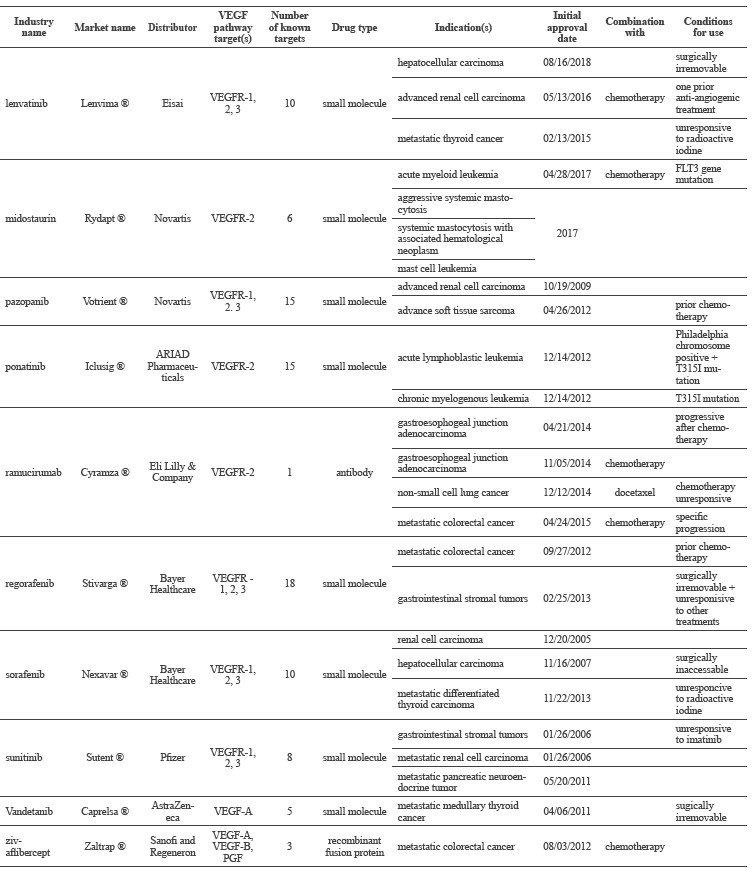
To conclude, thirteen drugs currently inhibit some part of the VEGF-pathway of tumor angiogenesis and are FDA approved to treat tumors. Two of these drugs reduce the concentration of active VEGF produced by a tumor (Category 1: Anti-VEGF), and eleven of these medications interfere with the receptors that VEGF binds to and signals through (Category 2: VEGFR inhibitors). As of 2016, 24 drugs that target the VEGFA pathway in tumor angiogenesis were in Phase III of clinical trials (Ferrara and Adamis, 2016). Nearly all of these were in combination therapies with an already approved drug (see Ferrara and Adamis, 2016). Given the 20% success rate of Phase III trials (Smith, 2007), within the next 1-3 years, 4-5 new drugs might be available to treat tumors via inhibition of the VEGF pathway of tumor angiogenesis.
ALTERNATIVE TREATMENTS
As we analyze what is available for treatment options, how will treatments improve? How will 4-5 new combinations of the same thing address the cancer epidemic in the next 1-3 years? Will tumor angiogenesis treatment culminate with bevacizumab combination trials and treatments until enough small molecules are found to block everything associated with tumor angiogenesis? Is such a farfetched goal even possible, or likely to be safe? And lastly, why have small molecules gained so much popularity over antibodies when the first breakthrough was with an antibody?
Fundamentally, pharmaceutical companies and the patents and pathways that lead them to optimal profits predictably determine what comes next. For now, that appears to be combination therapies that rely on the efficacy of drugs already approved by the FDA, with a heavy focus on the use of bevacizumab in combination treatment with new small molecules
(Ferrara and Adamis, 2016). The realistic evidence is that VEGF anti-angiogenic therapies may never actually improve beyond these antibody antagonists and small molecule inhibitors. The advances already achieved may be the extent that humans are capable of tolerating. Or, conceivably, bevacizumab was just the first huge step forward in cancer treatment, and the medical establishment is wary of drifting away from this initial success. One thing seems obvious, the current batch of next-generation anti-angiogenesis drugs are incremental, rather than offering the potential leap that led to the innovative treatments currently available today.
Proceeding beyond the confines of these current combination therapies will likely depend on the continued advancement of mechanistic understanding of all tumor angiogenesis pathways and the definitive processes that drive cells to become cancerous. Additionally, we need to understand better the angiogenic processes that remain obscure – the mechanisms and cascades of sprout elongation.
Combinations of small molecules very well may be the answer to treating cancer, but the present reality is that most of the combination therapies work as redundancies that target different steps in the same pathway, not as dual treatments of the various sources of angiogenesis; also, off-target binding causes complications. Nonetheless, combination treatments may help combat the drug-resistance to anti-angiogenesis therapies (Folkman, 2007; Welti et al., 2013).
Several reasons have contributed to the increased use of small molecules over antibodies in anti-VEGF tumor angiogenesis treatment. First, small molecules can be analyzed in mass quantities using a variety of high-throughput technologies. Second, how to administer antibodies remains problematic, since proteins and peptides require specific routes of administration. Small molecules also are not targeted by the immune system, but instead are physiologically filtered out of a body, whereas the immune system targets foreign antibodies “and may limit their utility” (R. Cohen, personal communication). Additionally, antibodies are more difficult and expensive to produce, and a challenge to construct, as obtaining the cells to make antibodies requires many methodical steps. Even after all that effort, there is no guarantee the antibodies will show any efficacy in pre-clinical phases, or that pre-clinical phases will correlate to clinical trials.
Alternative approaches in the literature
Two alternative therapies offer potential for targeting tumor angiogenesis with therapeutics – targeting DLL4-expressing stalk cells using conjugated peptides or using anti-DLL4 antibodies. In addition, a novel proposal for treating tumors utilizes components of the complement cascade and membrane attack complex.
DLL4 small molecule targeting (nanomedicine Drug Delivery Systems). The first potential treatment utilizes a nanomedicine drug delivery system (nDDS) to increase efficacy. Liu et al. (2015) adopted a drug delivery method to target currently approved drugs at the DLL4 membrane-bound ligand. They conjugated a 16-amino acid peptide to paclitaxel (a biologic that targets microtubules during cell division) to target the drug at DLL4 expressing cells in nude mice. The experiments showed excellent drug targeting to the tumor site, which contributed to improved anti-cancer drug efficacy in mice. These tests suggested that homing compounds and peptides may develop and evolve in synergy with drug compounds. Also, the research showed that the technology exists to reduce side effects and improve the efficacy of anti-angiogenesis drugs. The first potential treatment for future clinical use is the conjugation of targeting peptides to current drugs.
Anti-DLL4 antibody. The second potential treatment utilizes antibodies specific to DLL4 (anti-DLL4). OncoMed (Redwood City, CA) and Celgene (a subsidiary of Monsanto, St. Louis, MO) collaboratively developed an anti-DLL4 antibody known as demcizumab (OMP-21M18). Demcizumab is an antibody that competes with Notch receptors for DLL4 ligands. The DLL4 membrane-bound ligand has been a promising prospective target since scientists began to elucidate the tip cell-selecting role of DLL4 in tumor angiogenesis. The theory is that not allowing DLL4 to bind to Notch forces indiscriminate proliferation and undifferentiated selection of tip cells. This theory was proven correct in describing how anti-DLL4 antibodies function – by disrupting the dynamic balance between tip cell selection and stalk cell selection (Couch et al., 2016). Via this disruption, anti-DLL4 antibodies have shown considerable antitumor activity in preclinical research (Couch et al., 2016). Unfortunately, extrapolation of preclinical research to pharmacological treatment is not simple – the systemic effects of using anti-DLL4 antibodies have included adverse liver alterations, asymptomatic hypertension in humans, and vascular neoplasms upon prolonged exposure in lower mammals (Couch et al., 2016). In an attempt to counter the side effects of anti-DLL4 antibodies, Genentech began engineering F(ab’)2 regions of anti-DLL4 antibodies. The engineered F(ab’)2 regions promoted filtration of the antibody and, therefore, potentially reduced the side effects associated with cytotoxic antibodies circulating systemically for prolonged periods (Couch et al., 2016).
While the Genentech engineered antibody is still at the preclinical stage, OncoMed discontinued three clinical trials evaluating the anti-DLL4 antibody demcizumab in tumor therapy after combination studies with other drugs in first-line metastatic cancer (OncoMed, 2017).
Other angiogenesis-associated targets. Possible options for untargeted participants of the VEGF pathway of angiogenesis discussed in the literature include Notch receptors exclusive to angiogenesis, NICD, NECD, and VEGFR-1. There is no apparent evidence of any antibodies or small molecules being targeted at the Notch receptors, NICD, or NECD corresponding to stalk cells. The lack of targeting to Notch, NICD, and NECD is likely due to many off-target effects, indicated by the Notch pathway’s involvement in other physiological processes, including cell-fate determination in embryogenesis, physiologic development, epithelial tissue self-repair, and cardiac inflammation (Liu et al., 2014; Kandel et al., 2011; Quillard and Charreau, 2013). Scientists would first need to discover a Notch receptor isoform exclusive to tumor angiogenesis, before efficiently targeting Notch or its functional domains. Conclusively, there remain four obvious VEGF-pathway targets that have yet to receive attention in research and the literature. The four VEGF-pathway associated-targets are a Notch receptor exclusive to angiogenesis, NICD, NECD, and selective targeting of VEGFR-1.
Novel approach. A novel approach utilizes the complement cascade and resultant membrane attack complex – like Type-II hypersensitivity reactions. This review extends the above overview to propose a possible approach to VEGF-pathway targeted anti-angiogenesis tumor treatment using antibody-engineering techniques in concert with complement components of the immune system.
In the 1970s, a technique known as immunosurgery, in which the innate immune system is directed to destroy trophoblast and trophectoderm layers enveloping cell masses, came into prominence (Solter and Knowles, 1975; Handyside and Barton, 1977). This method currently allows embryonic stem cell harvesting from mammalian embryos (Tanaka et al., 2005; Chen and Melton, 2007). Immunosurgery engineers antibodies to target specific cells, and then induces the complement cascade system to destroy the targeted cells, while leaving non-targeted cells alone. In the case of harvesting stem cells, this involves using rabbit anti-human antibodies to target trophoblasts, and guinea pig serum complement to destroy the trophoblasts and expose the blastocyst cavity and inner cell mass of mammalian embryos. In the case of tumors, scientists can extrapolate immunosurgery to use antibodies to target the complement cascade at tip cells, proliferative stalk cells, or both.
Antibody-mediated cytotoxicity is already a means to target tumor cells directly (Taylor and Lindorfer, 2016; Stasiłojć et al., 2016), “as is amply demonstrated through studies of the anti-HER2 humanized antibody trastuzumab and others” (R. Cohen, personal communication; Cohen, 2012). However, by using the same technique – of engineered antibodies with altered Fc and Fab regions, to both induce cytotoxicity and increase the rate of elimination – against the proliferating blood supply, a growing tumor could hypothetically be asphyxiated as a whole, rather than eliminated cell-by-cell. For tip cell targeting, the cytotoxic antibodies would conceivably be anti-DLL4. For stalk cell targeting, the cytotoxic antibodies would conceivably be anti-Notch or anti-VEGFR-1. The nDDS homing technique could even be used to improve cytotoxic targeting. Exclusive targeting of tumor angiogenesis may someday be possible; this would allow the development of a broader range of anti-angiogenesis drugs, and enable the refinement of current drug concepts. The proposition is, therefore, to destroy – rather than neutralize – the cells responsible for vessel elongation. The membrane attack complex of the complement system targeting membrane-bound receptors of tip and stalk cells mediate the destruction.
CONCLUSION
The primary pathway of tumor angiogenesis is considered to be the VEGF-pathway; as such, it has become the leading target for pharmacological treatment of tumors. In a summary of tumor angiogenesis and the primary pharmaceutical targets within tumor angiogenesis: a tumor releases VEGF, VEGF binds to VEGFR-2,-3, VEGFR-2,-3 induces expression of DLL4, DLL4 binds to Notch, NICD is cleaved from NECD, and NICD induces expression of VEGFR-1. Research has shown that targeting this pathway destabilizes and inhibits vascular growth (angiogenesis) into a tumor.
Angiogenesis inhibitor treatments are most promising in preclinical research when they are applied before a tumor has become metastatic – that is before the tumor ceases to produce pro-angiogenic proteins (Folkman, 2003). In contrast, the thirteen VEGF-pathway angiogenesis inhibitor medications currently approved by the FDA are almost exclusively limited to forms of cancer that have already metastasized. Current angiogenesis inhibitors do not alter any established vasculature into a tumor – beyond reducing vascular leakiness (Folkman, 2007). As such, a dilemma emerges – angiogenesis inhibitors potently suppress tumor angiogenesis, but only reliably so if they are used to prevent a newly immortal tumor from gaining vasculature. In parallel to this dilemma, the current success of angiogenesis inhibitors, such as bevacizumab, in metastatic tumors, is likely due to the systemic repression or inhibition of migrating metastatic cells, and not due to vasculature prevention. The systemic inhibition prevents migrating metastatic cells from becoming resident at distant sites and allows for other drugs and treatments to target the tumor. Although these preventative techniques help restrict the further progression of tumors, they do not theoretically utilize VEGF anti-angiogenesis drug in an optimal way nor at the optimal time.
Future angiogenesis inhibitor research should focus on screening and detection efforts. Improved tumor detection technology would allow current angiogenesis inhibitor treatments to be delivered at the phases of tumor development when they can reach maximum efficacy. Although still useful in assisting in the treatment of metastatic tumors, angiogenesis inhibitors could be much more efficient as an adjuvant treatment targeted to hypoxic sites, to asphyxiated and kill immortal cells in primary tumors before they develop metastatic capabilities. Angiogenesis aimed immunosurgery could be a way to alter the established vasculature of metastatic tumors. In combination treatment with anti-VEGF agents and small molecules, immunosurgery could prove to be a defense against metastatic cells leaving a tumor, and a choke point from which to siege a tumor, as targeting both the endothelial cell and the cancer cell is more likely to succeed (Folkman, 2006). As our understanding of anti-angiogenesis therapy grows, it supports Cohen’s 2012 conclusion that:
The next phase in the war on cancer will be far more complex than the last, necessarily fragmented by new ways of interrogating and classifying tumors, and fueled by a plethora of new targeted agents. A new and re-invigorated approach to adjuvant trials will help ensure success (Cohen, 2012).
ACKNOWLEDGEMENTS
David J. Carr, M.D.; Kathleen M. Cohen, R.N.; Robert L. Cohen, M.D. - Subject Matter Expert; William DuMonthier, D.C.; William F. Jackson, Ph.D.; Mary M. Maison, M.P.A.; Richard B. Maison, M.P.A.; Amir J. Pakray, M.D., M.S.; Rosser Panggat, M.D.; Roseann L. Vorce, Ph.D.;
REFERENCES
Barbie, D.A., and Frank, D.A. 2012. Pharmacology of cancer signal transduction. In: Golan, D.E., Tashjian Jr., A.H., Armstrong, E.J., and Armstrong, A.W. Principle of pharmacology: the pathophysiologic basis of drug therapy. Lippincott Williams & Wilkins, a Wolters Kluwer business, Philadelphia. p. 699-715.
Boareto, M., Jolly, M.K., Ben-Jacob, E., and Onuchic, J.N. 2015. Jagged mediates differences in normal and tumor angiogenesis by affective tip-stalk fate decision. PNAS. 112: E-3836-E3844. https://doi.org/10.1073/pnas.1511814112
Chen, A.E., and Melton, D.A. 2007. Derivation of human embryonic stem cells by immunosurgery. Journal of Visualized Experiments. 10: 574. https://doi.org/10.3791/574
Cohen, R. L. 2012. Adjuvant trials of targeted agents: the newest battleground in the war on cancer. Current Topics in Microbiology and Immunology. 355: 217-232. https://doi.org/10.1007/82_2011_166
Couch, J.A., Zhang, G., Beyer, J.C., de Zafra, C.L., Gupta, P., Kamath, A.V., Lewin-Koh, N., Tarrant, J., Allamneni, K.P., Cain, G., et al. 2016. Balancing efficacy and safety of an anti-DLL4 antibody through pharmacokinetic modulation. Clinical Cancer Research. 22: 1469-1479. https://doi.org/10.1158/1078-0432.CCR-15-1380.
de Lartigue, J. 2016. VEGF remains central target for anti-angiogenic therapy despite challenges. Retrieved from http://www.onclive.com/publications/oncology-live/2016/vol-17-no-7/vegf-remains-central-target-for-antiangiogenic-therapy-despite-challenges/1
De Smet, F., Segura, I., De Bock, K., Hohensinner, P.J., and Carmeliet, P. 2009. Mechanisms of vessel branching. Arteriosclerosis, Thrombosis, and Vascular Biology. 29: 639-649. https://doi.org/10.1161/ATVBAHA.109.185165
El-Kenawi, A.E., and El-Remessy, A.B. 2013. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. British Journal of Pharmacology. 170: 712-729. https://doi.org/10.1111/bph.12344
Fang, S., and Salvan, P. 2011. Stem cells in tumor angiogenesis. Journal of Molecular and Cellular Cardiology. 50: 290-295. https://doi.org/10.1016/j.yjmcc.2010.10.024
Ferrara, N., and Adamis, A.P. 2016. Ten years of anti-vascular endothelial growth factor therapy. Nature Reviews Drug Discovery. 15: 385-403. https://doi.org/10.1038/nrd.2015.17
Ferrara, N., Hillan, K.J., Gerber, H.P., and Novotny, W. 2004. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nature Reviews Drug Discovery. 3: 391-400. https://doi.org/10.1038/nrd1381
Ferrara, N., Hillan, K.J., and Novotny, W. 2005. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochemical and Biophysical Research Communications. 333: 328-335. https://doi.org/10.1016/j.bbrc.2005.05.132
Folkman, J., and Shing, Y. 1992. Angiogenesis. Journal of Biological Chemistry. 267(16): 10931-4.
Folkman J. 1971. Tumor angiogenesis: therapeutic implications. New England Journal of Medicine. 285: 1182-1186. https://doi.org/10.1056/NEJM197111182852108
Folkman J. 2003. Angiogenesis inhibitors: a new class of drugs. Cancer Biology & Therapy. 2: S127-133
Folkman, J., and Kalluri, R. 2004. Cancer without disease. Nature. 427: 787. https://doi.org/10.1038/427787a
Folkman, J. 2006. Angiogenesis. Annual Review of Medicine. 57:1-18. https://doi.org/10.1146/annurev.med.57.121304.131306
Folkman J. 2007. Angiogenesis: an organizing principle for drug discovery? Nature Reviews Drug Discovery. 6: 273-286. https://doi.org/10.1038/nrd2115
Food and Drug Administration. 2017. Resources for information on approved drugs. Retrieved from https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm
Gerhardt, H., Golding, M., Fruttiger, M., Ruhrberg, C., Lundkvist, A., Abramsson, A., Jeltsch, M., Alitalo, K., Shima, D., and Betsholtz, C. 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. Journal of Cell Biology. 161: 1163-1177. https://doi.org/10.1083/jcb.200302047.
Handyside, A.H., and Barton, S.C. 1977. Evaluation of the technique of immunosurgery for the isolation of inner cell masses from mouse blastocysts. Journal of Embryology and Experimental Morphology. 37: 217-226.
Holmes, K., Roberts, O.L., Thomas, A.M., and Cross, M.J. 2007. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19: 2003-2012. https://doi.org/10.1016/j.cellsig.2007.05.013
Jain, R.K. 2003. Molecular regulation of vessel maturation. Natural Medicine. 9: 685-693. https://doi.org/10.1038/nm0603-685
Kandel, J.J., Yamashiro, D.J., and Kitajewski, J. 2011. Angiogenesis in tumor development and metastasis. In: Slevin, M. Therapeutic angiogenesis for vascular diseases: molecular mechanisms and targeted clinical approaches for the treatment of angiogenic disease. Springer Publishing, New York City. p. 81-93.
Kareva, I. 2016. Escape from tumor dormancy and time to angiogenic switch as mitigated by tumor-induced stimulation of stroma. Journal of Theoretical Biology. 395: 11-22. https://doi.org/10.1016/j.jtbi.2016.01.024
Leung, D.W., Cachianes, G., Kuang, W.J., and Ferrara, N. 1989. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 246: 1306-1309.
Liu, Y.R., Guan, Y.Y., Luan, X., Lu, Q., Wang, C., Liu, H.J., Gao, Y.G., Yang, S.C., Dong, X., Chen, H.Z., et al. 2015. Delta-like ligand 4-targeted nanomedicine for antiangiogenic cancer therapy. Biomaterials. 42: 161-171. https://doi.org/10.1016/j.biomaterials.2014.11.039
Liu, Z., Fan, F., Wang, A., Zheng, S., and Lu, Y. 2014. Dll4-Notch signalling in regulation of tumor angiogenesis. Journal of Cancer Research and Clinical Oncology. 140: 525-536. https://doi.org/10.1007/s00432-013-1534-x
Lodish, H., Berk, A., Kaiser, C.A., Krieger, M., Scott, M.P., Bretscher, A., Ploegh, H., and Matsudiara, P. 2007.Cancer. In: Lodish, H., Berk, A., Kaiser, C.A., Krieger, M., Scott, M.P., Bretscher, A., Ploegh, H., and Matsudaira, P. Molecular Cell Biology, W.H. Freeman and Company, New York. p. 1107-1150.
Mayor, R., and Etienne-Manneville, S. 2016. The front and rear of collective cell migration. Nature Reviews Molecular Cell Biology. 17: 97-109. https://doi.org/10.1038/nrm.2015.14.
Melero-Martin, J.M., and Dudley, A.C. 2011. Concise review: Vascular stem cells and tumor angiogenesis. Stem Cells. 29: 163-168. https://doi.org/10.1002/stem.583
National Cancer Institute. 2018. A to Z list of cancer drugs. Retrieved from http://www.cancer.gov/about-cancer/treatment/drugs
OncoMed. 2017. OncoMed’s Phase 2 Demcizumab Pancreatic Cancer Trial Misses Primary Endpoint. Retrieved from http://www.oncomed.com/invest/releasedetail.cfm?ReleaseID=1020677
Patel, N.S., Li, J.L., Generali, D., Poulsom, R., Cranston, D.W., and Harris, A.L. 2005. Upregulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Research. 65: 8690-8697. https://doi.org/10.1158/0008-5472.CAN-05-1208
Quillard, T., and Charreau, B. 2013. Impact of notch signaling on inflammatory responses in cardiovascular disorders. International Journal of Molecular Sciences. 14: 6863-6888. https://doi.org/10.3390/ijms14046863
Ridgeway, J., Zhang, G., Wu, Y., Stawicki, S., Liang, W., Chanthery, Y., Kowalski, J., Watts, R.J., Callahan, C., Kasman, I., et al. 2006. Inhibitors of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 444: 1083-1087. https://doi.org/10.1038/nature05313
Smith, L.J. 2007. Types of clinical studies. In: Evans, R. P. Drug and biological development: from molecule to product and beyond. Springer, New York. p. 107-122.
Solter, D., and Knowles, B.B. 1975. Immunosurgery of mouse blastocyst. Proceedings of the National Academy of Sciences. 72: 5099-5102.
Stasiłojć, G., Österborg, A., Blom, A.M., and Okrój, M. 2016. New perspective on complement mediated immunotherapy. Cancer Treatment Reviews. 45: 68-75. https://doi.org/10.1016/j.ctrv.2016.02.009
Tahergorabi, Z., and Khazaei, M. 2012. A review on angiogenesis and its assays. Iranian Journal of Basic Medical Sciences. 15(6): 1110-1126.
Tanaka, N., Takeuchi, T., Neri, Q.V., Rosenwaks, Z., and Palermo, G.D. 2005. Effect of immunosurgery on ICM attachment and embryonic stem cell harvesting. Fertility and Sterility. 84: S401-S401. https://doi.org/10.1016/j.fertnstert.2005.07.1048
Taylor, R.P., and Lindorfer, M.A. 2016. Cytotoxic mechanisms of immunotherapy: harnessing complement in the action of anti-tumor monoclonal antibodies. Seminars in Immunolog. 3: 309-16. https://doi.org/10.1016/j.smim.2016.03.003
Welti, J., Loges, S., Dimmeler, S., and Carmeliet, P. 2013. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. Journal of Clinical Investigation. 123: 3190-3200. https://doi.org/10.1172/JC170212
Wishart, D.S., Feunang, Y.D., Guo, A.C., Lo, E.J., Marcu, A., Grant, J.R., Sajed, T., Joshnson, D., Li, C., Sayeeda, Z., et al. 2018. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research. 46(D1): D1074-D1082. https://doi.org/10.1093/nar/gkx1037
World Health Organization. 2018. Cancer fact sheet. Retrieved from http://www.who.int/mediacentre/factsheets/fs297/en/
David P. Maison
1 Life Chiropractic College West, Hayward, California 94545, United States
2 Michigan State University, College of Osteopathic Medicine-Pharmacology & Toxicology, East Lansing, Michigan 48824-1316, United States
Corresponding author. E-mail: maisondp@gmail.com
Total Article Views
Article History:
Received: December 8, 2017
Revised: April 10, 2018
Accepted: April 19, 2018