 ISSN: 2822-0838 Online
ISSN: 2822-0838 Online

Hb Constant Spring and Its Combinations with Other Forms of Thalassemia or Hemoglobinopathy in Northern Thailand
Sopitnapa Kaewko, Chedtapak Ruengdit, Manoo Punyamung, and Sakorn Pornprasert*Published Date : September 22, 2025
DOI : https://doi.org/10.12982/NLSC.2026.001
Journal Issues : Number 1, January-March 2026
Abstract Hemoglobin Constant Spring (Hb CS; HBA2: c.427T>C) is a prevalent non-deletional α-thalassemia mutation frequently observed in Southeast Asia, particularly in Thailand. Given its clinical severity, especially when homozygous (αCSα/αCSα) or coinheritance with α⁰-thalassemia (αCSα /--) called Hb H-CS disease. Thus, early and accurate detection of Hb CS carriers is critical. However, routine laboratory screening can be challenging due to the low levels of Hb CS in heterozygous individuals. This study aimed to examine Hb CS and its combinations with other forms of thalassemia or hemoglobinopathies in Northern Thailand using a targeted next-generation sequencing (NGS). Hemoglobin analysis was routinely performed using high-performance liquid chromatography (HPLC) and/or capillary electrophoresis (CE) techniques. The α0-thalassemia --SEA, --THAI, and --Chiang Rai type deletions were identified using real-time polymerase chain reaction (PCR) combined with high-resolution melting (HRM) analysis. The Hb CS mutation was identified in 147 out of 2,773 samples (5.3%). The study consisted of 17 distinct genotypes in which revealed variability in clinical and hematological characteristics based on their combinations. Therefore, the identification of Hb CS is certainly useful in thalassemia screening, genetic counselling, control, prevention, and treatment.
Keywords: Hb constant spring, Hematological parameters, Hemoglobinopathy, Next-generation sequencing, Thalassemia
Graphical Abstract:

Citation: Kaewko, S., Ruengdit, C., Punyamung, M., and Pornprasert, S. 2026. Hb constant spring and its combinations with other forms of thalassemia or hemoglobinopathy in Northern Thailand. Natural and Life Sciences Communications. 25(1): e2026001.
INTRODUCTION
Hb Constant Spring (Hb CS; HBA2: c.427T>C) is one of the most prevalent non-deletional α-thalassemia mutations. This mutation results from a single nucleotide substitution at the termination codon of the HBA2 gene. It leads to the production of an elongated α-globin chain with an additional 31 amino acids (Liao et al., 2010; Waneesorn et al., 2011; Pornprasert et al., 2012). The elongation of the α-globin chain leads to mRNA instability. Consequently, the synthesis of the αCS-globin chain is reduced (Bunn and Forget, 1986). In Southeast Asia, the gene frequency of Hb CS ranges from 3% to 6% (Uaprasert et al., 2011). In Thailand, the mutation is particularly prevalent in the northeastern regions, where its frequency varies between 1% and 8% (Fucharoen and Winichagoon, 1987; Laig et al., 1990; Jomoui et al., 2015). In contrast, in Northern Thailand, it is reported at approximately 3.3% (Laig et al., 1990). Homozygosity for Hb CS or compound heterozygosity for Hb CS and α0-thalassemia named Hb H-CS disease, can result in severe fetal anemia, which triggers hydrops fetalis due to the fetus's organs being unable to regulate the anemia, causing widespread fluid buildup and potentially stillbirth. This occurs because the mutated Hb CS gene results in an unstable or less functional hemoglobin, impairing oxygen delivery to tissues and overwhelming the fetal circulatory system. This often requires life-saving intrauterine transfusions (Sirilert et al., 2019; Luewan et al., 2020). In 2006, the first documented case of hydrops fetalis associated with homozygosity for Hb CS was successfully managed with intrauterine blood transfusion (Charoenkwan et al., 2006). HbH disease is classified into deletional and non-deletional types. Deletional HbH disease occurs when both α-globin gene copies on one chromosome 16 are deleted and a single α-globin gene is deleted on another chromosome 16 (α-/--). In contrast, non-deletional HbH disease results from point mutations affecting one α-globin gene combined with two α-globin gene deletions on another chromosome 16 (αTα /--) (Chui et al., 2003). A notable example of a non-deletional mutation is Hb CS, which disrupts the termination codon and produces an unstable elongated α-globin chain (Lal et al., 2024). In both HbH (α-/--) and Hb H-CS (αCSα /--) diseases, the predominant pathogenic mechanism is the accumulation of excess β-globin chains within erythroid cells. The resultant β-globin homotetramer (HbH; β4) is unstable but remains more structurally intact than excess α-globin chains in β-thalassemia, leading to reduced ineffective erythropoiesis (Schrier et al., 1997; Pootrakul et al., 2000). Ferrokinetic studies show that hemolysis is the main pathological feature in HbH disease rather than ineffective erythropoiesis. In contrast, HbH-CS disease has significantly increased ineffective erythropoiesis due to the instability of elongated αCS-globin chains, which precipitate in the bone marrow and cause oxidative stress, damaging erythroid progenitor cells (Schrier et al., 1997; Pootrakul et al., 2000). Given the clinical severity of such cases, early identification of Hb CS carriers among parents is crucial to assess fetal risk and enable timely medical interventions. The mRNA of the Hb CS mutant gene is highly unstable. This leads to a reduced rate of α-chain synthesis and results in low levels of Hb CS in peripheral blood, making detection particularly challenging in heterozygous individuals (Bunn and Forget, 1986). This study aims to use the next-generation sequencing (NGS) panel designed to target the coding regions of the HBA1, HBA2, and HBB genes for the precise diagnosis of Hb CS and its combinations with other forms of thalassemia or hemoglobinopathies in Northern Thailand. Moreover, the association between genotypes and phenotypes of each combination form was also described.
MATERIAL AND METHODS
This research received ethical approval from the Ethics Committee at the Faculty of Associated Medical Sciences, Chiang Mai University, Thailand (Approval No. AMSEC-67EM-037). Blood samples anti-coagulated with ethylenediamine tetra acetic acid (EDTA) and the data of red cell indices, including the red blood cell counts (RBCs), total Hb, packed cell volume (PCV), mean corpuscular volume (MCV), mean corpuscular Hb (MCH), and mean corpuscular Hb concentration (MCHC) measured by automated cell counters were submitted from public and private hospitals in Northern Thailand to the Associated Medical Sciences-Clinical Service Center (AMS-CSC), Chiang Mai University, Chiang Mai, Thailand for routine thalassemia diagnosis. At the AMS-CSC, a high-performance liquid chromatography (HPLC) (VARIANT β-thalassemia Short Program; Bio-Rad Laboratories, Hercules, CA, USA) and/or a capillary electrophoresis (CE) system (Capillarys 2 Flex piercing, Sebia, Evry, France) were used to quantify HbA2 (for β-thalassemia detection) and to identify hemoglobinopathy. DNA extraction from EDTA whole blood samples was performed using the Chelex method (Chelex 100 Resin, Sigma, CA, USA (Walsh et al., 1991). The α0-thalassemia diagnosis specifically for --SEA, --THAI, and --Chiang Rai deletions was performed using single-tube multiplex real-time PCR combined with EvaGreen dye and high resolution melting (HRM) analysis, as previously described (Ruengdit et al., 2023). Additionally, other thalassemia mutations and hemoglobinopathies were identified using the NGS panel designed to target the coding regions of the HBA1, HBA2, and HBB genes. This assay utilized the Thalassemia Gene Detection Kit provided by BGI Group (Shenzhen, China). Subsequently, sequence reads were mapped against the human genome reference hg19, and detected genetic variants were analyzed using THACARE HALOS software (BGI Group) (He et al., 2017; Li et al., 2021).
RESULTS & DISCUSSION
From August to November 2023, a total of 2,773 blood samples underwent globin gene variant screening using NGS. The sequencing result of Hb CS identified by NGS is presented in Figure 1. Based on the NGS system and routine thalassemia diagnosis methods, the Hb CS mutation was identified in 147 individuals (5.3%), including 5 and 142 samples with homozygosity and heterozygosity for Hb CS, respectively. Thus, the Hb CS gene frequency in our studied population was 0.027. Consistency with the previous study revealed that the Hb CS gene frequency was 0.033 in Northern Thailand (Laig et al., 1990). Moreover, the combinations of Hb CS with other forms of thalassemia or hemoglobinopathy could be classified into 17 distinct genotypes. The characteristics and hematological parameters of these 17 genotypes are summarized in Table 1. The heterozygosity for Hb CS demonstrated variable MCV and MCH levels ranging from slightly below the normal ranges to normal values. Total Hb levels generally remained within normal limits (Table 1), suggesting mild or no anemia. Importantly, conventional Hb analysis methods, such as the CE system, failed to detect the Hb CS peak (Figure 2A), highlighting potential diagnostic limitations as observed in the previous study (Jaitheang et al., 2023). Consequently, heterozygosity for Hb CS could be misclassified, posing a risk in genetic counselling. For instance, parents may be unaware of their carrier status, which increases the likelihood of having offspring with severe forms of thalassemia such as Hb H-CS disease or homozygosity for Hb CS, which can lead to hydrops fetalis (Chui et al., 2003; Charoenkwan et al., 2006; Lal et al., 2024). Previous studies demonstrated that the CE method outperforms HPLC for detecting Hb CS, with 92.3-100% of samples exhibiting heterozygosity for Hb CS identified by CE, compared to only 26.3-48.2% detected by HPLC. Furthermore, all samples exhibiting homozygosity for Hb CS were identified by CE, whereas detection rates by HPLC ranged from 42.9% to 100% (Waneesorn et al., 2011; Embong et al., 2024).
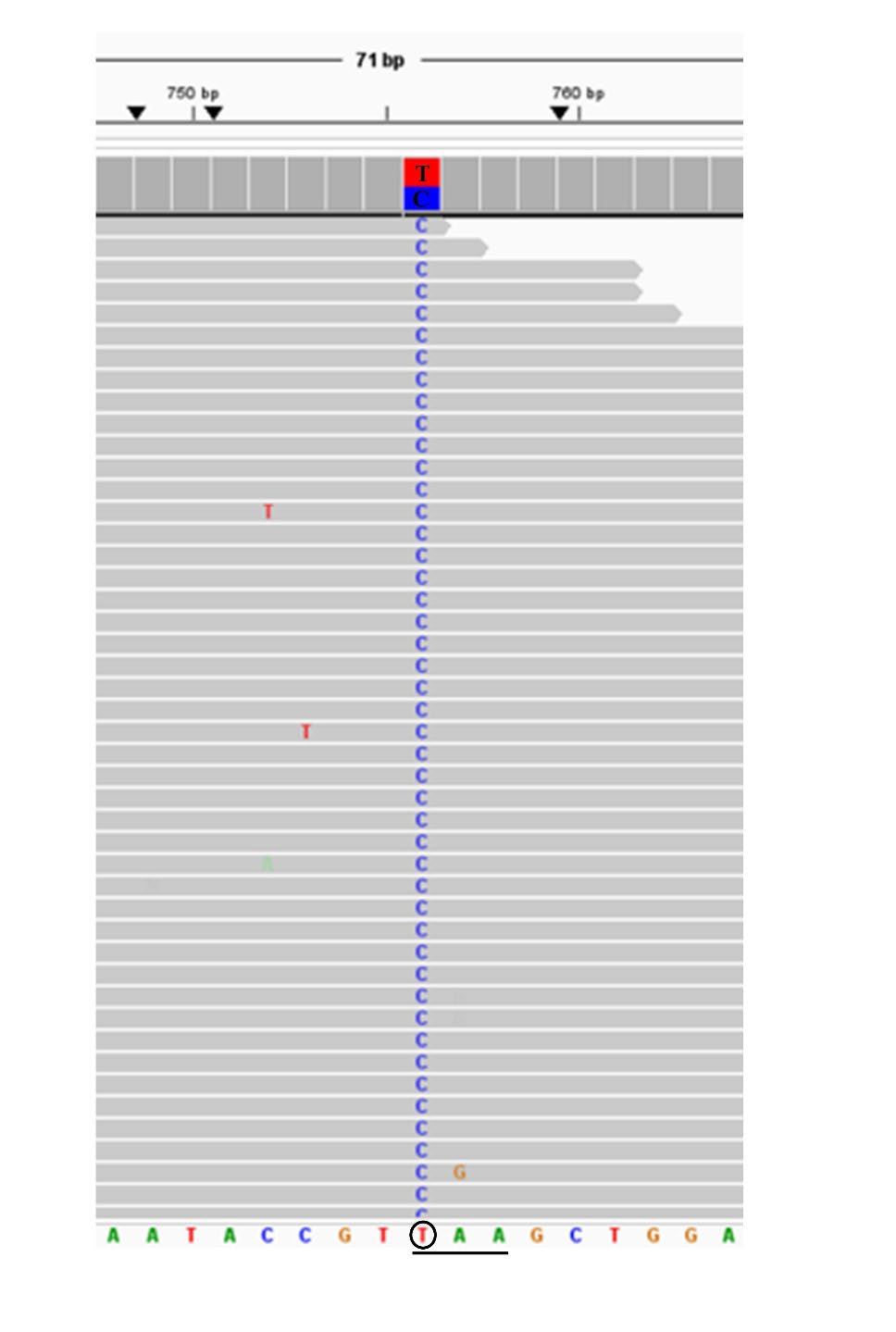
Figure 1. Representative of NGS results exported from the integrative genomics viewer (IGV) of the heterozygosity for Hb CS (HBA2: c.427T>C).
Double heterozygosity for Hb CS and β+- or β0-thalassemia presented lower MCV and MCH values than heterozygosity for Hb CS. It also showed elevated HbA2 (>3.5%) and mild anemia (Table 1). The CE electropherograms (Figure 2B) and HPLC chromatograms (Figure 2C) failed to identify the Hb CS peak. This heightened the likelihood of misdiagnosis. Consistent with the prior study, the CE method was unable to identify the Hb CS individuals who had double heterozygosity for Hb CS and β-thalassemia. Even though, the CE method offers significantly higher resolution than traditional screening techniques for detecting Hb CS, the amount of Hb CS (α2CSβ2) may be too low in these subjects due to reduced β-globin chain expression (Li et al., 2013).
Table 1. Characteristics, hematological parameters, and hemoglobin analysis of Hb Constant Spring and its combinations.
|
Table 1. Characteristics, hematological parameters, and hemoglobin analysis of Hb Constant Spring and its combinations. |
|
||||||||||
|
Genotypes (N; Male/Female) |
Age (Years) (Ranges) |
Hematological parameters |
Hb analysis by HPLC or CE (%) |
||||||||
|
RBCs (x1012/L) |
Total Hb (g/dL) |
PCV (L/L) |
MCV (fL) |
MCH (pg) |
MCHC (g/L) |
HbA |
HbA2 |
HbF |
|||
|
αα/αCSα, bA/bA (55; 20/35) |
40.1 ± 19.2 (1.0 – 82.0) |
4.4 ± 1.0 (1.8 – 6.6) |
11.1 ± 2.5 (3.8 – 17.4) |
0.34 ± 0.70 (0.13 – 0.51) |
77.6 ± 7.3 (60.0 – 95.0) |
25.0 ± 2.8 (16.6 – 30.6) |
322.3 ± 15.8 (276.2 – 358.0) |
89.1 ± 12.9 (7.4 – 98.3) |
2.4 ± 0.5 (1.5 – 3.3) |
0.6 ± 0.9 (0.0 – 4.1) |
|
|
αα/αCSα, b+/bA (1; 1/0) |
1.0 |
5.5 |
10.6 |
0.34 |
62.0 |
19.2 |
309.9 |
85.6 |
4.7 |
9.7 |
|
|
αα/αCSα, b0/bA (1; 0/1) |
19.0 |
3.8 |
9.0 |
0.28 |
73.0 |
23.7 |
324.9 |
80.4 |
6.6 |
6.7 |
|
|
αα/αCSα, bE/bA (16; 7/9) |
39.4 ± 16.7 (20.0 – 73.0)
|
4.6 ± 0.9 (2.9 – 6.0) |
11.7 ± 2.5 (6.9 – 15.7) |
0.36 ± 0.07 (0.22 – 0.46) |
77.0 ± 3.3 (70.0 – 83.0) |
25.1 ± 1.2 (23.1 – 26.5) |
326.6 ± 9.4 (300 – 341) |
69.5 ± 6.6 (58.6 – 77.6) |
*25.6 ± 2.0 (22.4 – 29.9) |
0.6 ± 0.7 (0.0 – 2.4) |
|
|
αα/αCSα, bE/bE (1; 1/0) |
62.0 |
4.1 |
8.9 |
0.26 |
64.0 |
21.9 |
342.3 |
0.0 |
*100.0 |
0.0 |
|
|
αCSα/αCSα, bA/bA (4; 1/3) |
59.7 ± 29.9 (26.0 – 83.0)
|
4.1 ± 0.7 (3.3 – 4.7) |
9.4 ± 1.1 (8.6 – 10.6) |
0.32 ± 0.03 (0.29 – 0.35) |
77.3 ± 8.5 (71 – 87) |
23.0 ± 2.6 (20.6 – 25.8) |
297.7 ± 8.1 (290.3 – 306.4) |
87.4 ± 4.1 (82.7 – 92.6) |
2.4 ± 0.8 (1.4 – 3.3) |
1.2 ± 0.6 (0.5 – 1.7) |
|
|
αCSα/αCSα, b0/bA (1; 0/1) |
37.0 |
5.3 |
11.1 |
0.37 |
70.0 |
21.0 |
300.0 |
93.1 |
3.9 |
0.0 |
|
|
αCSα/--SEA, bA/bA (20;13/7) |
21.7 ± 20.0 (1.0 – 64.0)
|
4.0 ± 1.0 (2.0 – 5.9) |
7.7 ± 2.2 (3.7 – 12.8) |
0.27 ± 0.07 (0.12 – 0.38) |
67.5 ± 6.8 (56.0 – 82.0) |
19.2 ± 2.7 (15.3 – 27.3) |
283.8 ± 18.3 (260.9 – 332.5) |
85.9 ± 7.3 (72.2 – 96.0) |
1.3 ± 1.0 (0.4 – 3.3) |
0.9 ± 0.9 (0.0 – 3.1) |
|
|
αCSα /--SEA, b0/bA (2;2/0) |
1.0 & 64.0
|
6.2 & 4.1 |
8.7 & 6.7
|
0.29 & 0.24
|
47.0 & 59.0 |
14.1 & 16.5 |
300.0 & 279.2 |
92.0 & 88.0 |
2.2 & 4.5 |
5.1 & 0.8 |
|
|
αCSα /--SEA, bE/bA (4;2/2) |
23.2 ± 29.4 (6.0 – 67.0)
|
4.4 ± 1.0 (3.3 – 5.4) |
7.5 ± 0.6 (6.9 – 8.0) |
0.26 ± 0.01 (0.25 – 0.26) |
60.3 ± 15.7 (48.0 – 78.0) |
17.9 ± 5.3 (14.4 – 24.0) |
294.4 ± 16.5 (276.0 – 307.7) |
76.8 ± 9.6 (63.2 – 83.8) |
*17.8 ± 6.5 (12.2 – 27.2) |
1.0 ± 0.9 (0.0 – 2.1) |
|
|
-α3.7/αCSα, bA/bA (28;12/16) |
34.4 ± 24.0 (1.0 – 87.0)
|
4.9 ± 1.3 (1.2 – 6.6) |
10.7 ± 2.4 (3.9 – 13.7) |
0.34 ± 0.07 (0.13 – 0.43) |
72.0 ± 10.1 (64.0 – 108.0) |
22.5 ± 3.1 (19.3 – 32.4) |
312.0 ± 15.6 (294.6 – 361.8) |
93.6 ± 5.1 (81.0 – 98.4) |
2.2 ± 0.4 (1.6 – 3.0) |
0.4 ± 0.6 (0.0 – 2.5) |
|
|
-α4.2/αCSα, bA/bA (2;0/2) |
0.3 & 0.3 |
4.5 & 4.5 |
10.6 & 10.8 |
0.34 & 0.34 |
76.0 & 76.0 |
23.7 & 24.1 |
311.8 & 317.6 |
77.9 & 78.0 |
2.1 & 2.0 |
13.7 & 13.6 |
|
|
-α3.7/αCSα, bE/bA (7;1/6) |
25.4 ± 16.6 (1.0 – 40.0)
|
5.0 ± 0.3 (4.6 – 5.4) |
9.8 ± 2.7 (4.7 – 11.9) |
0.33 ± 0.04 (0.27 – 0.38) |
65.0 ± 9.4 (50.0 – 74.0) |
19.5 ± 5.6 (8.7 – 23.4) |
294.7 ± 59.3 (174.1 – 326.2) |
76.6 ± 7.8 (63.2 – 82.6) |
*18.2 ± 1.4 (15.7 – 19.2) |
3.5 ± 8.1 (0.0 – 20.0) |
|
|
αWMα/αCSα, bA/bA (2; 1/1) |
15.0 & 21.0
|
4.1 & 5.4 |
11.2 & 14.2 |
33.0 & 42.8 |
80.0 & 79.0 |
27.2 & 26.2 |
339.4 & 331.8 |
96.3 & 88.4 |
2.4 & 3.2 |
0.6 & 0.7 |
|
|
αIVSI-1α/αCSα, bA/bA (1; 0/1) |
59.0 |
5.1 |
7.9 |
0.27 |
53.0 |
15.5 |
292.6 |
98.1 |
1.9 |
0.0 |
|
|
αIVSI-1α/αCSα, b0/bA (1; 0/1) |
17.0 |
4.3 |
9.3 |
0.30 |
69.0 |
21.4 |
310.0 |
84.8 |
5.5 |
2.2 |
|
|
αCSα/αααanti-4.2, bE/bA (1; 0/1) |
18.0 |
2.4 |
6.4 |
0.21 |
86 |
26.2 |
304.8 |
76.4 |
*23.6 |
0.0 |
|
|
Note: Normal range: red blood cell counts (RBCs), 4.2 - 6.1x1012/L; total Hb, 12.0 -18.0 g/dL; packed cell volume (PCV), 0.37 - 0.52 L/L; mean corpuscular volume (MCV), 80 - 100 fL; mean corpuscular Hb (MCH), 27 - 31 pg; mean corpuscular Hb concentration (MCHC), 320 - 360 g/L; HbA >85%; HbA2 1.5 - 3.5%; HbF 0.0 - 1.0%, *HbA2 = HbA2 + HbE |
|||||||||||
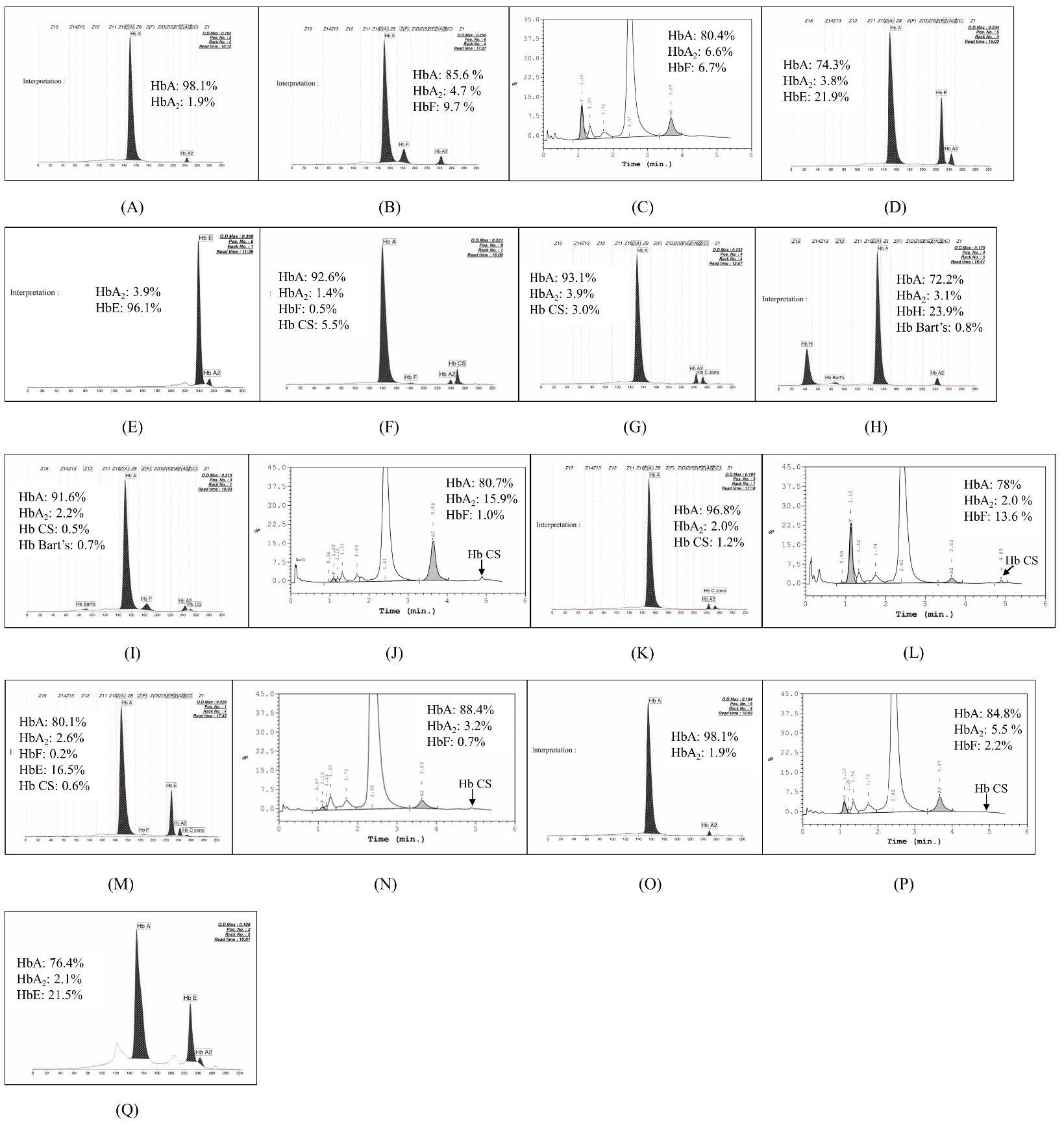
Figure 2. Representative of The HPLC chromatogram and/or CE electropherogram of 17 genotypes, including (A) αα/αCSα, βA/ βA (A), αα/αCSα , β+/βA (B), αα/αCSα , β0/ βA (C), αα/αCSα, βE/βA (D), αα/αCSα, βE/βE (E), αCSα/αCSα, βA/βA (F), αCSα/αCSα, β0/βA (G), αCSα/--SEA, βA/βA (H), αCSα/--SEA, β0/βA (I), αCSα/--SEA, βE/βA (J), -α3.7/αCSα, βA/βA (K), -α4.2/αCSα, βA/βA (L), -α3.7/αCSα, βE/βA (M), αWMα/αCSα, βA /βA (N), α IVS-I-1α/αCSα, βA/βA (O), αIVS-I-1α/αCSα, β0/βA (P), and αCSα/αααanti-4.2, β0/βA (Q).
Double heterozygosity for Hb CS and HbE demonstrated similarly reduced MCV and MCH values as the combinations with β-thalassemia. The HbA2 + HbE levels notably exceeded 25% due to the presence of HbE. The severity of anemia varied significantly among these individuals (Table 1). The CE electropherogram consistently did not reveal the Hb CS peak (Figure 2D). In contrast, the previous study showed that Hb CS can be detected by the CE system in all subjects with double heterozygosity for Hb CS and HbE (Pornprasert et al., 2015). The co-inheritance of heterozygosity for Hb CS and homozygosity for HbE led to reduced MCV and MCH in comparison to double heterozygosity for Hb CS and HbE, while HbA2 + HbE levels attained 100% (Table 1). The CE electropherogram consistently failed to detect Hb CS (Figure 2E) which potentially misclassified these samples as solely homozygosity for HbE.
The homozygosity for Hb CS exhibited slightly reduced MCV and MCH with mild anemia (Table 1). The CE electropherogram clearly identified the Hb CS peak which was distinctively higher than those seen in other genotype groups (Figure 2F). In addition, the co-inheritance of homozygous for Hb CS and heterozygous β0-thalassemia displayed lower MCV and MCH values compared to homozygosity for Hb CS alone but had improved total Hb levels (Table 1). The CE electropherogram showed lower Hb CS peak (Figure 2G) compared to homozygosity for Hb CS who had no β-thalassemia mutations, likely due to decreased β-globin synthesis. This resulted in reduced binding of β-globin chain to αCS-globin chain hence lowering Hb CS (α2CSβ2). Mild anemia, characterized by a mean Hb level of 10 g/dL, had previously been observed in 22 individuals with homozygosity for Hb CS, aged 7 to 42 years. Nevertheless, certain individuals in this cohort exhibited mild hepatosplenomegaly and jaundice (Pootrakul et al., 1981). Furthermore, Komvilaisak et al. (2018) shown that homozygosity for Hb CS could result in moderate to severe hemolytic anemia, requiring intense phototherapy and blood transfusions throughout fetal and neonatal periods due to anemia and jaundice. However, mild hemolytic anemia may occur in adults without requiring transfusion due to elevated HbA and reduced HbF levels compared to fetal and neonatal stages. Viprakasit et al. (2004) reported a case of a 7-year-old male with homozygosity for Hb CS, who had severe hemolytic anemia following a febrile episode triggered by a bacterial or viral infection. Thus, these results indicated that persons with homozygosity for Hb CS typically exhibit mild symptoms unless provoked by an external stimulus as they age.
Patients with Hb H-CS (αCSα/--SEA) showed reduced MCV and MCH values and severe anemia (Table 1). Some required regular transfusions. The CE electropherogram (Figure 2H) displayed the peaks of HbH and Hb Bart’s. Most cases showed detectable Hb CS peak. However, certain individuals lacked this peak, and this could potentially result in a misdiagnosis as deletional HbH disease. Consistency with the previous study showed that compound heterozygosity for Hb CS and α0-thalassemia (αCSα/--SEA) results in a form of non-deletional Hb H-CS disease that is more clinically severe than the triple α-globin gene deletion called deletional HbH disease (-α/--SEA). Patients with this disease have lower baseline Hb and some are even transfusion dependent (He et al., 2015). In the current study, there was a patient with Hb H-CS disease whose total Hb was 3.7 g/dL (Table 1) and required a blood transfusion.
The co-inheritance of Hb H-CS disease and β0-thalassemia showed severely reduced MCV and MCH values (Table 1). HbA2 levels typically exceeded 3.5%. Although one child (1 year old) had a lower level of HbA2 (2.2%), it was likely due to incomplete d-chain synthesis at this age. The total Hb levels remained low and comparable to those observed in Hb H-CS disease without co-inheritance of β-thalassemia. The CE electropherogram (Figure 2I) displayed the Hb CS peak without detectable HbH because the reduced β-globin production leads to a decrease in HbH which consists of four β-globin chains. The co-inheritance of Hb H-CS disease and heterozygosity for HbE had lower MCV and MCH values compared to the Hb H-CS disease alone and more severe anemia (Table 1). HbA2 + HbE levels remained below 25%. The HPLC chromatogram (Figure 2J) showed Hb CS peaks without detectable HbH. The reasons underlying this are similar to those when it comes to Hb H-CS disease, who is inherited with β0-thalassemia.
Subjects with compound heterozygosity for Hb CS and α⁺-thalassemia (-α3.7 or -α4.2) showed slight reductions in MCV and MCH along with mild anemia (Table 1). Both CE electropherogram (Figure 2K) and HPLC chromatogram (Figure 2L) identified the Hb CS peaks. Consistency with the prior study indicated that hematological parameters, including total Hb, MCV, MCH, and MCHC, in individuals with compound heterozygosity for Hb CS and α⁺-thalassemia were significantly lower than those in individuals with heterozygosity for Hb CS alone. In addition, Hb CS levels of compound heterozygosity for Hb CS and α⁺-thalassemia were significantly higher than those of heterozygosity for Hb CS alone (Uaprasert et al., 2011). Subjects with compound heterozygosity for Hb CS and α⁺-thalassemia and HbE trait had lower levels of MCV and MCH compared to those without HbE trait (Table 1). Due to the reduced imbalance of β- and α-globin ratio, HbA2 + HbE levels remained below 25%. The CE electropherogram revealed the Hb CS peak (Figure 2M).
Other genotype combinations, including αWMα/αCSα, αIVSI-1 α/αCSα, αIVSI-1 α/αCSα combined with β0-thalassemia, and αCSα/αααanti-4.2 combined with HbE were observed in the current study. The αWMα/αCSα individuals generally had normal hematological values (Table 1). Consistency with the previous study showed that the RBC indices were mildly affected by heterozygosity for Hb Westmead (Jiang et al., 2020) but could present mild anemia, with detectable Hb CS peaks by HPLC (Figure 2N). The αIVSI-1α/αCSα patients had more severe anemia compared to the compound heterozygosity for α⁺-thalassemia and Hb CS (Table 1). This was possibly exacerbated by the IVSI-1 mutation. No Hb CS peaks were observed via CE electropherogram (Figure 2O). It should be noted that the αIVSI-1(AGGT > AGAT) mutation (IVSI-1, HBA1: c.95 + 1G > A) is an α+-thalassemia determinant caused by transition of G > A of the donor splice consensus site sequence of the first intron of the α1-globin gene (Harteveld et al., 2000). The combination of αIVSI-1α/αCSα with β0-thalassemia alleviated anemia severity due to a balance of α- and β-globin ratio, with a tiny peak of Hb CS on HPLC chromatogram (Figure 2P). Finally, the αCSα /αααanti-4.2 combined with HbE trait showed normal MCV and MCH (Table 1) due to the balance of α-globin synthesis as the presence of an extra α-gene alleviates the imbalance caused by Hb CS. However, reduced Hb levels were possibly due to decreased RBC production. The Hb CS peak was not detected by CE system (Figure 2Q). Contrary to the previous study showed that triplicated or quadruplicated α-globin genes increase the severity of HbE syndromes (Vichinsky, 2007). The limitations of the present study are some genotypes have hematological parameters derived from only 1-2 individuals thus, further studies are required. Furthermore, other etiologies of microcytic anemia, including iron deficiency and lead poisoning, could not be excluded.
CONCLUSION
The frequency of Hb CS in Northern Thailand was 5.3% and it could be co-inherited with other thalassemia or hemoglobinopathies, resulting in a total of 17 different genotypes. The clinical and hematological features varied with their combination forms. These demonstrations will enable genetic counseling, support prevention, and control programs for thalassemia and hemoglobinopathy not only in Northern Thailand but also in Asian countries where high frequency of these hematological disorders. Several molecular techniques, such as the Amplification Refractory Mutation System (ARMS) and Real-time PCR with High-Resolution Melting (HRM) analysis, can be employed for the detection of Hb CS (Li et al., 2010; Waneesorn et al., 2011). However, the NGS panel targeting the coding regions of HBA1, HBA2, and HBB genes is more effective than these approaches for assessing the genotype and phenotype associations of Hb CS and its interactions with other forms of thalassemia or hemoglobinopathy.
ACKNOWLEDGMENTS
The authors thank technicians in the Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand for their help and assistance.
AUTHOR CONTRIBUTIONS
SK, CR, MP, and SP were involved in the conception of the study. SK and CR performed the data collection and the analyses. SK, CR, and SP were involved in the interpretation of the results. SK and SP drafted the manuscript. CR and MP gave critical feedback on the manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
CONFLICT OF INTEREST
The authors declare that they hold no competing interests.
REFERENCES
Bunn, H.F. and Forget, B.G. 1986. Hemoglobin: Molecular, genetic and clinical aspects. New York: W B Saunders Co.
Charoenkwan, P., Sirichotiyakul, S., Chanprapaph, P., Tongprasert, F., Taweephol, R., Sae-Tung, R., and Sanguansermsri, T. 2006. Anemia and hydrops in a fetus with homozygous hemoglobin constant spring. Journal of Pediatric Hematology/Oncology. 28(12): 827-830.
Chui, D.H., Fucharoen, S., and Chan, V. 2003. Hemoglobin H disease: Not necessarily a benign disorder. Blood. 101(3): 791-800. https://doi.org/10.1182/blood-2002-07-1975
Embong, S.F., Daud, A., Nordin, M.H., and Adzahar, S. 2024. Detection of hemoglobin constant spring: A comparison of capillary electrophoresis versus high-performance liquid chromatography. Cureus. 16(8): e67228. https://doi.org/10.7759/cureus.67228
Fucharoen, S. and Winichagoon, P. 1987. Hemoglobinopathies in southeast Asia. Hemoglobin. 11(1): 65-88. https://doi.org/10.3109/03630268709036587
Harteveld, C., Beijer, C., Van Delft, P., Zanardini, R., Bernini, L., and Giordano, P. 2000. α‐Thalassaemia as a result of a novel splice donor site mutation of the α1‐globin gene. British Journal of Haematology. 110(3): 694-698. https://doi.org/10.1046/j.1365-2141.2000.02225.x
He, J., Song, W., Yang, J., Lu, S., Yuan, Y., Guo, J., Zeng, X., Zhang, Y., Peng, M., Quang, H.H., et al. 2017. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: The Dai nationality, China. Genetics in Medicine. 19(9): 1022-1031. https://doi.org/10.1038/gim.2016.218
He, S., Zheng, C., Meng, D., Chen, R., Zhang, Q., Tian, X., and Chen, S. 2015. Hb H hydrops fetalis syndrome caused by association of the --(SEA) deletion and Hb constant spring (HBA2: c.427T>C) mutation in a Chinese family. Hemoglobin. 39(3): 216-219. https://doi.org/10.3109/03630269.2015.1030031
Jaitheang, J., Suksusut, A., Settapiboon, R., Amornsiriwat, S., Sutcharitchan, P., Uaprasert, N., and Rojnuckarin, P. 2023. Stability of Hemoglobin constant spring identified by capillary electrophoresis. Laboratory Medicine. 54(3): e91-e94. https://doi.org/10.1093/labmed/lmac130
Jiang, F., Ju, A.P., Li, J., Chen, G.L., Zhou, J.Y., Tang, X.W., Zuo L.D., and Li, D.Z 2020. Hb westmead (HBA2: c.369C>G): Hematological characteristics in heterozygotes with and without α0-thalassemia. Hemoglobin. 44(3): 153-155. https://doi.org/10.1080/03630269.2020.1768109
Jomoui, W., Fucharoen, G., Sanchaisuriya, K., Nguyen, V.H., and Fucharoen, S. 2015. Hemoglobin constant spring among Southeast Asian populations: Haplotypic heterogeneities and phylogenetic analysis. PLoS One. 10(12): e0145230. https://doi.org/10.1371/journal.pone.0145230
Laig, M., Pape, M., Hundrieser, J., Flatz, G., Sanguansermsri, T., Das, B., Bernini, L., Giordano, P., Harteveld, C., Zanardini, R., et al. 1990. The distribution of the Hb constant spring gene in Southeast Asian populations. Human Genetics. 84(2): 188-190. https://doi.org/10.1007/BF00208939
Lal, A., Viprakasit, V., Vichinsky, E., Lai, Y., Lu, M.Y., and Kattamis, A. 2024. Disease burden, management strategies, and unmet needs in α‐thalassemia due to hemoglobin H disease. American Journal of Hematology. 99(11): 2164-2177. https://doi.org/10.1002/ajh.27440
Li, R., Liao, C., Li, D., and Li, J. 2010. High-resolution melting analysis of the three common nondeletional α-thalassemia mutations in the Chinese population: Hbs Constant Spring, Quong Sze and Westmead. Hemoglobin. 34(6): 587-593. https://doi.org/10.3109/03630269.2010.526881
Li, R., Shen, X., Chen, H., Peng, D., Wu, R., and Sun, H. 2021. Developmental validation of the MGIEasy Signature Identification Library Prep Kit, an all-in-one multiplex system for forensic applications. International Journal of Legal Medicine. 135(3): 739-753. https://doi.org/10.1007/s00414-021-02507-0
Li, Y.Q., Li, R., and Li, D.Z. 2013. Detection of Hb constant spring [α142, Term→ Gln, TAA > CAA (α2)] in heterozygotes combined with β-thalassemia. Hemoglobin. 37(2): 197-200. https://doi.org/10.3109/03630269.2013.768532
Liao, C., Zhou, J.Y., Xie, X.M., Li, J., Li, R., and Li, D.Z. 2010. Detection of Hb constant spring by a capillary electrophoresis method. Hemoglobin. 34(2): 175-178. https://doi.org/10.3109/03630261003680191
Luewan, S., Charoenkwan, P., Sirichotiyakul, S., and Tongsong, T. 2020. Fetal haemoglobin H‐Constant Spring disease: A role for intrauterine management. British Journal of Haematology. 190(4): e233-e236. https://doi.org/10.1111/bjh.16809
Pootrakul, P., Sirankapracha, P., Hemsorach, S., Moungsub, W., Kumbunlue, R., Piangitjagum, A., Wasi, P., Ma, L., Schrier, S.L., et al. 2000. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia: Presented in part at the XXI congress of the international society of hematology, Sydney, Australia, may 1985, and the annual meeting of the American society of hematology, San Diego, CA, December 1997. Blood, The Journal of the American Society of Hematology. 96(7): 2606-2612. https://doi.org/10.1182/blood.V96.7.2606
Pootrakul, P., Winichagoon, P., Fucharoen, S., Pravatmuang, P., Piankijagum, A., and Wasi, P. 1981. Homozygous haemoglobin constant spring: A need for revision of concept. Human Genetics. 59(3): 250-255. https://doi.org/10.1007/BF00283674
Pornprasert, S., Panyasai, S., Waneesorn, J., Kongthai, K., and Singboottra, P. 2012. Quantification of hemoglobin constant spring in heterozygote and homozygote by a capillary electrophoresis method. International Journal of Laboratory Hematology. 34(2): 143-147. https://doi.org/10.1111/j.1751-553X.2011.01371.x
Pornprasert, S., Saoboontan, S., and Punyamung, M. 2015. Detection of Hb constant spring (HBA2: c.427T>C) heterozygotes in combination with β-thalassemia or Hb E trait by capillary electrophoresis. Hemoglobin. 39(3): 211-215. https://doi.org/10.3109/03630269.2015.1027827
Ruengdit, C., Punyamung, M., Intasai, N., and Pornprasert, S. 2023. Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of α0-thalassemia --SEA, --THAI, and --CR type deletions. PLoS One. 18(11): e0293838. https://doi.org/10.1371/journal.pone.0293838
Schrier, S., Bunyaratvej, A., Khuhapinant, A., Fucharoen, S., Aljurf, M., Snyder, L.M., Keifer, C.R., Ma, L., and Mohandas, N. 1997. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood. 89(5): 1762-1769. https://doi.org/10.1182/blood.V89.5.1762
Sirilert, S., Charoenkwan, P., Sirichotiyakul, S., Tongprasert, F., Srisupundit, K., Luewan, S., and Tongsong, T. 2019. Prenatal diagnosis and management of homozygous hemoglobin constant spring disease. Journal of Perinatology. 39(7): 927-933. https://doi.org/10.1038/s41372-019-0397-7
Uaprasert, N., Rojnuckarin, P., Settapiboon, R., Amornsiriwat, S., and Sutcharitchan, P. 2011. Hematological characteristics and effective screening for compound heterozygosity for Hb constant spring and deletional α+-thalassemia. American Journal of Hematology. 86(7): 615-617. https://doi.org/10.1002/ajh.22033
Vichinsky, E. 2007. Hemoglobin E syndromes. Hematology. 2007(1): 79-83. https://doi.org/10.1182/asheducation-2007.1.79
Walsh, P.S., Metzger, D.A., and Higuchi, R. 1991. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 10(4): 506-513.
Waneesorn, J., Panyasai, S., Kongthai, K., Singboottra, P., and Pornprasert, S. 2011. Comparison between capillary electrophoresis and high performance liquid chromatography for detection and quantification of Hb constant spring [Hb CS; α142, Term→ Gln (TAA> C AA IN α2)]. Hemoglobin. 35(4): 338-345. https://doi.org/10.3109/03630269.2011.588140
OPEN access freely available online
Natural and Life Sciences Communications
Chiang Mai University, Thailand. https://cmuj.cmu.ac.th
Sopitnapa Kaewko1, Chedtapak Ruengdit2, Manoo Punyamung2, and Sakorn Pornprasert1, *
1 Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
2 Associated Medical Sciences Clinical Service Center, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
Corresponding author: Sakorn Pornprasert, E-mail: sakornmi001@gmail.com; sakorn.pornprasert@cmu.ac.th
ORCID iD: Sakorn Pornprasert: https://orcid.org/0000-0003-0280-5569
Total Article Views
Editor: Decha Tamdee,
Chiang Mai University, Thailand
Article history:
Received: May 13, 2025;
Revised: August 27, 2025;
Accepted: August 29, 2025;
Online First: September 22, 2025